

Label The Drawing Of An Enteric Bacterium With The Antigens And Virulence Factors.
Introduction
Shigella was recognized as the causative agent of bacillary dysentery in 1897 by Kiyoshi Shiga. He determined that it was a Gram negative bacillus, which was capable of fermenting dextrose, but was indole-reaction negative and incapable of producing acid from mannitol (Trofa et al., 1999). Shigella is a non-sporulating, facultative anaerobe. Shigella is also a primate-restricted pathogen, which differentiates it from the other members of the Enterobacteriaceae family in which it is classified.
The Shigella genus is divided into four species: Shigella dysenteriae (serogroup A, 15 serotypes), Shigella flexneri (serogroup B, 19 serotypes), Shigella boydii (serogroup C, 20 serotypes), and Shigella sonnei (serogroup D, 1 serotype). These are divided into multiple serotypes dependent on O-antigen and biochemical differences. Different species are linked to disease in varying geographical locations. S. dysenteriae causes severe epidemic disease in less developed countries, S. flexneri causes disease in developing countries, S. boydii is confined to the Indian subcontinent, and S. sonnei occurs in both transitional and developed countries (Levine et al., 2013).
Shigellosis is the clinical presentation of Shigella infection. Disease is transmitted through the fecal-oral route, with an infectious dose of only 10–100 organisms (Levine et al., 2013). After 1–4 days, infection is acute, non-systemic and enterically invasive, leading to destruction of the colonic epithelium (detailed in Figure 1). Damage along the colonic epithelial is dramatic but erratic, and leads to the main clinical symptom of diarrhea, containing blood and sometimes mucus, which may be accompanied by abdominal cramps and fever. Further complications, depending on the infecting Shigella species and host HLA subtype, include Haemolytic-Uremic Syndrome (HUS) and Post-Reactive Arthritis (WHO, 2005). HUS occurs in 2–7% of S. dysenteriae type 1 infections, whereby the Shiga toxin harbored by this species attaches to the endothelium and activates platelets, which adhere to the endothelium and occlude blood vessels leading microangiopathic haemolysis of red blood cells as they squeeze through the restricted blood vessel lumen (O'Loughlin and Robins-Browne, 2001). Symptoms include acute renal failure, thrombocytopenia, micro-angiopathic haemolytic anemia, with a 35% fatality rate (Mayer et al., 2012). Post-reactive arthritis is another complication of Shigella infection, occurring in 2% of cases, and is characterized by painful joints, painful urination, and irritation of eyes, with chronic arthritis lasting from months to years.
Figure 1. Infectious cycle of Shigella (Roerich-Doenitz, 2013) modified from Schroeder and Hilbi (2008) . Entry into the colonic epithelium is mediated in two ways: M-cell membrane ruffling, and epithelial barrier destabilization. Entry via M-cells is achieved through membrane ruffling (1), and the bacillus is then transported to the M-cell pocket, where it is endocytosed by resident macrophages (2). Epithelial barrier destruction is mediated by pro-inflammatory (IL-1) and chemotaxic cytokines (IL-8). IL-8 produced by neighboring epithelial cells recruits PMN leukocytes (12), which travel from the basolateral to the apical colonic epithelium, destabilizing the junctions between the epithelial cells and allowing further invasion of Shigella (5). Induction of pyroptotic macrophage death occurs after Shigella escape from the phagocytic vacuole (3 and 6). Caspase-1, when activated, cleaves and activates IL-1β and IL-18, leading to the release of these pro-inflammatory cytokines (4) (Jennison and Verma, 2004). Uptake of Shigella is a macropinocytic process at the basolateral membrane of epithelial cells (7). Stimulation of Rho-family GTPases triggers actin polymerisation and then depolymerization, forming filopodial and lamellipodial extensions of the epithelial membrane, leading to engulfment of the bacilli. Lysis of the macropinocytic vacuole allows Shigella to gain access to the epithelial cytoplasm, where it rapidly multiplies, escapes autophagy and fragments the Golgi (8 and 9). Exploitation of the epithelial actin assembly machinery allows Shigella to move both intra- and intercellularly (10). Protrusions mediated by bacilli are actively endocytosed by the clathrin-mediated endocytic pathways at intercellular junctions, and the double membrane vacuole is lysed to give Shigella access to the neighboring cells cytoplasm (11). PMN leukocytes eventually eliminate Shigella infection from the colonic epithelium (13).
Comparison of the main subtypes of these species by Yang et al. (2005) indicates that each Shigella species contains a single circular chromosome and a virulence plasmid. The virulence plasmid has been thoroughly researched in relation to pathogenesis, and the majority of the important virulence factors involved in the Shigella life-cycle are localized to a 30 kb region termed the "entry region" (Figure 2). This region contains the mxi-spa locus, which encodes the Type III Secretion System (T3SS), and ipa and ipg genes, which are essential for invasion of epithelial cells and initiation of Shigella infection. In addition to the virulence plasmid, distinct regions within the Shigella chromosome have also been shown to contribute to infection. These are termed "pathogenicity islands" (PAI) (Table 1), which are unstable transferable elements that can be found in a variety of combinations depending on the Shigella species and subtype (Yang et al., 2005). A combination of both chromosomal virulence factors and plasmid virulence factors mediate the Shigella life cycle that leads to destruction of the colonic epithelium and disease symptoms.
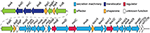
Figure 2. Genomic organization of the entry region on plasmid pWR100 (Roerich-Doenitz, 2013). Genes are clustered in two operons, the ipa/ipg and the mxi/spa operon. They are colored in the legend according to their protein class, some of which, such as T3SS effectors, are detailed in the text. Secretion machinery refers to the components that build the T3SS. Translocators are components of the translocon, a pore inserted into the host membrane to allow effector translocation and chaperones are components that stabilize individual effectors prior to secretion from the bacterium. Regulators modulate T3SS expression and function but they are largely beyond the scope of this review. This figure was modified from the virulence plasmid map by Buchrieser et al. (2000).
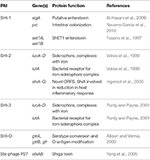
Table 1. Genes and protein functions involved in virulence on the Shigella chromosome.
The T3SS harbored by Shigella is pivotal to infection, delivering from the bacterial cytoplasm into the host cell effectors that play a role in cellular invasion, manipulation, and apoptosis (Parsot, 2009). At 37°C the T3SS components are assembled (Figure 3) but secretion of effectors is prevented until the T3SS is activated by contact with the host cell (Veenendaal et al., 2007). These effectors can be classified dependent on the timing of gene expression as either early, middle, or late effectors, and unless stated otherwise, all of the effectors discussed below are encoded on the virulence plasmid and secreted in a T3SS-dependent manner (Table 2).
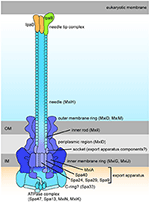
Figure 3. Schematic drawing of the Shigella Type III Secretion System (Roerich-Doenitz, 2013). The architecture of Shigella T3SS is based on the virulence plasmid mxi-spa locus (Figure 2), which encodes proteins that produce the T3SS apparatus, a structure composed of a 60 nm hollow extracellular needle, a transmembrane domain, and a cytoplasmic bulb (Blocker et al., 1999).
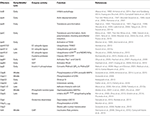
Table 2. Effectors secreted in a T3SS-dependent manner.
Dysentery, the main clinical symptom of shigellosis, is due to the infectious cycle of Shigella and its ability to penetrate and colonize the colonic epithelium, leading to loss of barrier function and inflammation (Jennison and Verma, 2004). This initial inflammation (Figure 1) is paramount for efficient infection. However, Shigella must also overcome this innate immune response and dampen inflammation in order to establish infection, especially in the epithelial cell niche. Shigella diminishes the inflammatory response by delivering effectors to inhibit the NFκB and MAPK signaling pathways and epigenetically regulate the repression of pro-inflammatory cytokines such as IL-8 (Ashida et al., 2011). In addition, Shigella is capable of downregulating production of antimicrobial peptides, including human β-defensin hBD-3, and chemokines, such as CCL20, leading to defective dendritic cell recruitment (Sperandio et al., 2008). This allows for increased replication, efficient infection of neighboring cells, and evasion of the immune response (Figure 1). Eventually, however, the initial inflammatory response which allows for efficient infection consequently leads to Shigella clearance. Polymorphonuclear leukocytes (PMN), such as neutrophils, eliminate the infection within 5–7 days in healthy individuals (Figure 1).
Shigella has evolved to successfully re-infect its host and probably subverts the production of efficient immunological memory to do so. After infection, seroconversion produces protective antibodies against Shigella lipopolysaccharide (LPS), however the antibodies produced are serogroup specific. The diversity of Shigella LPS serotypes means that protection against re-infection is limited to homologous disease, and these LPS-specific antibodies are also short lasting (Cohen et al., 1991). Progressive acquisition of pan-species immunological memory occurs after many infections, and is probably achieved through recognition of protein-based specific antigens, such as the "invasion plasmid antigens" (Ipa) proteins. These virulence plasmid-encoded antigens are therefore important targets for vaccine development (Levine et al., 2013). The adaptive immune response, including T- and B-lymphocytes, takes 4–7 days to begin working efficiently, which coincides with resolution of Shigella infection in healthy individuals. Shigella prevents dendritic cell recruitment by downregulating the main chemoattractant CCL20, as previously described, and also mediates dendritic cell apoptosis (Kim et al., 2008). Dendritic cells are a key link between the innate and adaptive immune response, and the inhibition of their function consequently interferes with the TH1-TH2-TH17 transition required for an efficient adaptive immune response (Sperandio et al., 2008). Therefore, it is possible that prevention of an efficient adaptive immune response is achieved via a combination of innate immune system modulation and subversion of immunological memory production.
The individual virulence factors of Shigella have been compiled and reviewed previously. Here we aim to understand how they collaborate to cause acute enteric destruction, leading to the clinical manifestation of shigellosis. We will analyse the available primary data for the function of Shigella effector proteins and their effect on host cells, and then discuss how these are co-ordinated in time and space to create a detailed picture of Shigella infection, how it leads to disease and manipulates the immune response.
Analysis
Epithelial Barrier Destabilization and Inflammation
OspB: Promotes PMN Migration, Inflammation, and Cell Proliferation
Like most effectors, OspB is found in the four Shigella species, and has homolog in Salmonella species (Zurawski et al., 2009). Although its biochemical function is unknown, it is thought that OspB plays a role in the activation of extracellular-signal-regulated kinases (ERK) and p38 MAPK pathways, resulting in phosphorylation of phospholipase A2 and the generation of eicosanoids. OspB is capable of nuclear localization for activation of MAPK signaling pathways. This contributes to inflammation and PMN migration, possibly inducing hepoxilin A3, an arachidonic acid derivative, and apical secretion of IL-8, a PMN chemoattractant (Ambrosi et al., 2014). An ospB − mutant had a 60% decrease in PMN migration and a 30% decrease in ERK1/2 activation 90 min post-infection when compared to wild-type Shigella (Zurawski et al., 2009). Furthermore, Ambrosi et al. (2014) showed that an ospB − knockout displayed significantly reduced onset and severity of symptoms in the guinea pig keratoconjunctivitis model of infection (Sereny test). However, OspB also activates the master regulator of cell growth mTOR via a direct interaction with the cellular scaffold protein IQGAP1, which also interacts with mTOR activators ERK1/2. This seems to restricts the spread of S. flexneri in cell monolayers, possibly by enhancing cell proliferation in infected foci (Lu et al., 2015).
OspC1: Promotes PMN Migration and Inflammation
OspC1 is part of the ospC family. There is 96% identity between ospC2, ospC3, and ospC4, but only 74% identity between these three ospC genes and ospC1 (Buchrieser et al., 2000). This level of similarity may indicate redundancy. However, ospC4 is a pseudogene and different functions have been identified for OspC1 and OspC3 (discussed later). Tagged OspC1 is found throughout the host cytoplasm, localizing primarily to the nucleus (Zurawski et al., 2006). An ospC1 − knockout showed a significant decrease in the amount of neutrophil recruitment to the epithelial cells in PMN migration assays, which was restored to wild-type levels on complementation with a plasmid expressing ospC1. Zurawski et al. (2006) showed that this increase in PMN migration correlated with increase in the phosphorylation of ERK1/2 pathways mediated by OspC1. An ospC1 − knockout showed a decrease in phosphorylation of ERK1/2 compared to wild-type levels but no reduction in IL-8 secretion. OspC1 plays a role in Shigella virulence in vivo as an ospC1 − knockout had reduced amounts of swelling and inflammation in the Sereny test, with clearance of infection after 2 days (Zurawski et al., 2006).
OspZ: Promotes PMN Migration and Inflammation
In S. flexneri 2a, an ospZ − knockout has no effect on the Sereny test. However, an ospZ − knockout caused a significant decrease in PMN migration. The knockout also had 63 and 53% ERK1/2 phosphorylation and NFκB activation, respectively, when compared to wild-type S. flexneri (Zurawski et al., 2008). OspZ therefore plays a role in the migration of PMN leukocytes across the epithelial barrier. However, Newton et al. (2010) discovered that S. flexneri species, excluding S. flexneri serotype 6, contain a stop codon at amino acid 188, forming a truncated protein lacking an IDSYMK motif at position 209. The full length OspZ proteins in the remaining Shigella species were found to have an immunosuppressive function through prevention of NFκB activation. Finally, an OspZ homolog, NleE, is found in enteropathogenic Escherichia coli (EPEC), and both NleE and OspZ can substitute for each other (Zurawski et al., 2008).
Serine Protease Autotransporters of Enterobacteriaceae
Serine Protease Autotransporters of Enterobacteriaceae (SPATEs) are a family of proteases which catalyse their own secretion via the Type V secretion pathway. Shigella has three known SPATEs, not all of which are found in each species. Their secretion is thermoregulated (37°C) and pH-dependent (Dautin, 2010). They have different proposed activities relevant to intestinal penetration: induction of mucin secretion and cleavage (Pic), destabilization of focal adhesions via cleavage of fodrin (SigA), and, through unknown targets, enterotoxicity, fluid accumulation and epithelial desquamation (SigA and SepA) (Table 3).

Table 3. SPATEs harbored by Shigella and their function in infection.
Shigella Enterotoxin 1 and Shigella Enterotoxin 2: Enterotoxic Activity in the Jejunum
Shigella enterotoxin 1 (ShET1) and Shigella enterotoxin 2 (ShET2) are virulence determinants proposed to mediate early fluid secretion in the jejunum to establish infection in the colon and produce to the characteristic watery diarrhea seen early in shigellosis. The shared name is due to their similar properties as enterotoxins, as there is no homology between ShET1 and ShET2.
ShET1 is encoded by set1A and set1B genes on the Shigella chromosome as part of the SHI-1 PAI, and only present in S. flexneri 2a isolates (Vargas et al., 1999) (Yavzori et al., 2002). The two subunits are proposed to form a holo-AB-type toxin complex in an A1-B5 configuration, producing a 55 kDa complex (Fasano et al., 1995). The holotoxin may follow a secretion mechanism similar to that of the cholera holotoxin, via the Sec pathway and Type II secretion. When ion transport across a cultured epithelium was measured in an Using chamber, a set1AB − knockout had 60% lower Isc (short circuit current) in comparison to wild-type strains (Faherty et al., 2012b). The effect on Isc of ShET1 was also dose-dependent, and washout of ShET1 produced no change in Isc, indicating irreversible binding of ShET1 to epithelial receptors (Fasano et al., 1997). However, the set1A and set1B genes overlap with the pic gene but are divergently transcribed. Therefore, the additional pic − knockout may have caused these effects. Faherty et al. (2012b) complemented the pic/set1AB mutant with pic and set1AB individually, showing that pic has a more significant contribution to restoring Isc levels to wild-type, although set1AB complementation also produced a significant increase in Isc.
ShET2 is a 63 kDa protein encoded by ospD3 (sen). It is found in all serotypes and one of the three ospD genes found on the virulence plasmid. Sequence alignments between ospD2 and ospD3 show a high degree homology while ospD1 is more divergent. OspD1 has a unique role in regulating type III secretion not shared with OspD2 and OspD3 (Parsot et al., 2005), but redundancy in their effector function(s) is unknown. Unlike ShET1, ShET2 secretion is dependent on the T3SS (Farfán et al., 2011) but how this is regulated unclear (Faherty et al., 2016). The ospD3 − (ShET2) knockout has similar effects in Using chamber experiments to a set1AB − (ShET1) knockout, with reduced Isc increase in comparison to the wild-type strain (Nataro et al., 1995). An ospD3 − mutant also had a reduction in IL-8 secretion, which could be restored to wild-type levels by ospD3 plasmid complementation, indicating a possible role in IL-8 secretion by epithelial cells (Farfán et al., 2011).
Adhesion to the Colonic Epithelium at the Basolateral Surface
Lipopolysaccharide: Glucosylation for T3SS Accessibility
The lipopolysaccharide (LPS) is a common feature of Gram negative pathogens, triggering the host immune response and inflammatory reactions during infection. LPS modification by glucosylation is thought to contribute to Shigella adhesion and invasion by revealing the T3SS for efficient activation upon contact with the host cell. Guan et al. (1999) showed that glycosyltransferase gtrA − and gtrB − mutants had only a partial conversion of the O-antigen serotype, and a gtrX − mutant had no conversion at all. A mutation in the gtr operon leads to a reduced ability to invade, and this invasion is restored when the gtr operon is reintroduced (West et al., 2005). The reduction in O-antigen length by glucosylation enhances accessibility of the T3SS for contact with the host epithelial cell to initiate invasion.
IpaB: Binds CD44 at the Basolateral Surface
IpaB mediates adhesion to the basolateral membrane via interactions with the ubiquitous glycoprotein CD44 (Figure 1, step 7). CD44 is located within lipid microdomain rafts, and is involved in binding of ezrin, radixin, and moesin (ERM) proteins to produce rearrangements of the actin cytoskeleton. IpaB binds the CD44 N-terminal domain with weak affinity but up-regulation of CD44 expression to levels found on lipid microdomains increases binding and internalization of Shigella (Skoudy et al., 2000). Lipid microdomain rafts are found at the basolateral surface and the IpaB-CD44 adhesion interaction may contribute to the polarity of Shigella invasion of epithelial cells (Lafont et al., 2002). Although increased adherence mediated by IpaB-CD44 binding may improve invasion efficiency, this binding alone is not sufficient to induce Shigella entry, as both ipaC − and ipaD − (see below) mutants are unable to mediate invasion (Skoudy et al., 2000).
IcsA (VirG): Polar Adhesion
IcsA, also referred to as VirG, is a 120 kDa outer membrane protein. IcsA is not dependent on the T3SS for its secretion as it is an autotransporter, with an atypical N-terminal signal sequence mediating secretion via the Sec pathway (Brandon et al., 2003). It is most well-known for its involvement actin based motility, however a more recent function has been described, whereby IcsA is involved in polar adhesion of Shigella to epithelial cells (Brotcke Zumsteg et al., 2014). The adhesion function can be separated from actin-based motility, as an icsA − mutant complemented with a plasmid encoding an adhesion-defective icsA formed plaques similar to wild-type Shigella (Brotcke Zumsteg et al., 2014). The adhesion-defective icsA also produced an attenuated infection phenotype in the Sereny test, indicating the importance of IcsA as an adhesin in Shigella pathogenesis. IcsA-mediated adhesion was present in an ipaBCDA − mxiE − mutant, but not in an ipaD − spa33 − strain, indicating that the assembled T3SS, but not the secretion of T3SS effectors, is required for adhesion activity. This initial observation was then linked to T3SS-dependent activation of IcsA to mediate this adhesive phenotype, as the application of the bile salt deoxycholate (DOC) led to an increase in IcsA-dependent adhesion (Brotcke Zumsteg et al., 2014). Deoxycholate has previously been described to bind to IpaD at the T3SS needle tip and induce IpaB recruitment for T3SS activation (Barta et al., 2012). However, DOC is also known to effect LPS molecules, causing them to disperse within the membrane (Shands and Chun, 1980). Assessment of protease accessibility using proteinase K with an ipaD − mutant and after DOC treatment showed that hyperadhesive IcsA was more resistant to degradation, and this was proposed to be due to an alternate conformation of IcsA induced by T3SS activation (Figures 4C, 5D—Brotcke Zumsteg et al., 2014). However, as DOC does not activate the T3SS, the changes in proteolysis banding are more likely due to disruption of the LPS leading to altered accessibility of IcsA producing different cleavage patterns. Changes seen in the ipaD − mutant could occur via an independent regulatory pathway, whereby activation of the T3SS leads to modulation of LPS structure.
OpsE1/E2: Bile Salts-Dependent Adhesion
Faherty et al. (2012a) also noticed that subculture in media containing bile salts significantly enhanced ability of Shigella to adhere to the apical surface of polarized epithelial cells. However, microarray expression analysis indicated that the ospE1/ospE2 genes were induced in the presence of bile, and bile-induced adherence was lost in a ΔospE1/ΔospE2 mutant. The OspE1/OspE2 proteins, which are effectors secreted by the T3SS, were also shown to remain localized to the bacterial outer membrane following exposure to bile salts, where they may therefore serve as adhesins.
Macropinocytic Uptake into Colonic Epithelial Cell
IpaD and IpaB: Activation of the T3SS
IpaD is part of the Ipa family required for Shigella invasion, and polymerizes at the T3SS needle tip (Espina et al., 2006). The identification of Class I ipaD − mutants, which had premature secretion of effectors, and Class II ipaD − mutants, which were non-inducible, shows that IpaD has a dual role in the activation of the T3SS (Roehrich-Doenitz et al., 2013). IpaD acts as the scaffold protein at the tip of the T3SS needle acting as the display support for IpaB, located at the needle tip along with IpaD (Veenendaal et al., 2007), and the delivery mechanism of the hydrophobic IpaB-IpaC translocation pore (translocon) to host cell membranes (Blocker et al., 1999). From there IpaD acts as a signal transducer to activate effector secretion (Figure 4) (Roehrich-Doenitz et al., 2013). IpaD, in conjunction with MxiC, is also part of the cytoplasmic signal transduction pathway required for full activation of the T3SS and secretion of remaining T3SS effectors (Martinez-Argudo and Blocker, 2010). IpaB initially senses the host cell membrane in a manner that is not yet understood and, with IpaD, co-transduces this signal down the T3SS needle to activate secretion (Murillo et al., 2016).
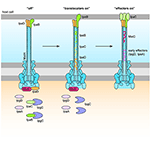
Figure 4. Role of IpaD, IpaB, and IpaC in regulation of the T3SS (Martinez-Argudo and Blocker, 2010). Four hydrophilic IpaD molecules and one hydrophobic IpaB molecule are localized at the tip of the T3SS needle in the "off" state. IpaB senses host cells upon contact of the T3SS needle tip, and inserts into the membrane to signal "translocators on." Secretion of effectors is signaled by conformational changes in IpaD via a signal transduction pathway to the base of the secreton. Four IpaC may travel up the secreton and associate with the needle tip, one atop each IpaD, and insert around the single IpaB to form a translocon in the epithelial cell membrane. This signals "effectors on" and early effectors are injected into the cytoplasm of the host epithelial cell (Veenendaal et al., 2007).
IpaC: Actin Polymerization and Induction of Effector Translocation
IpaC belongs to the group of Ipa proteins crucial for translocon formation and cell invasion. The structure of IpaC includes an N-terminal signal sequence, a region for association with IpgC (cytoplasmic chaperone), a central hydrophobic region for penetration of membranes, and a C-terminal domain for oligomerization (Terry et al., 2008). The hydrophobic region allows IpaC to interact with IpaB and insert into host membranes, however its topology in the membrane is disputed. The use of anti-IpaC monoclonal antibodies has shown that both the N- and C-terminal regions face the host cell cytoplasm (Tran Van Nhieu et al., 1999) although other experiments have found that the central loop is on the cytoplasmic face, with the N- and C-terminal regions being extracellular (Kuwae et al., 2001). IpaC insertion into the epithelial cell membrane triggers cytoskeletal rearrangements for the macropinocytic uptake of Shigella (Figure 4). Menard et al. (1996) describe how an IpaB-IpaC complex on the surface of latex beads is sufficient for engulfment by non-phagocytic cells. However, this has not been reproduced and therefore may not applicable in vivo. It is likely that the IpaC C-terminus faces the host cytoplasm because IpaC has been attributed the ability to polymerize actin indirectly at its C-terminus, via interactions with Cdc42 and Rac GTPases (Tran Van Nhieu et al., 1999) and activation of Src tyrosine kinase pathway (Mounier et al., 2009). However, the Salmonella homolog, SipC, can polymerize actin at its C-terminal domain, independently of any host cell factor (Hayward and Koronakis, 1999). IpaC displays sequence similarity to SipC within its C-terminal actin nucleation domain, and both IpaC and SipC use this domain to oligomerize. IpaC may therefore also be capable of polymerizing actin independently. In pull down assays, the C-terminal domain of IpaC was incapable of binding to Cdc42 and Rac1, indicating that actin polymerization does not occur through direct interactions with these GTPases (Terry et al., 2008). Finally, the C-terminus of IpaC has also been shown to bind vimentin and the intestinal epithelial intermediate filament keratin 18. This interaction is required for stable docking of the bacteria to cells and a prerequisite for induction of secretion of the other effectors (Russo et al., 2016).
IpgB1 and IpgB2: Actin Remodeling
IpgB1 and IpgB2 share 25% amino acid identity and both require Spa15 as a chaperone for secretion, with an additional requirement for stability by IpgB1 (Hachani et al., 2007). They contain a WxxxE motif which is common in guanine nucleotide exchange factors (GEFs) involved in the activation of Rho GTPases. These GTPases are required for the induction of actin filament structures to produce membrane ruffles for bacterial entry into non-phagocytic cells. An ipgB1 − knockout has a 50% reduction in epithelial cell invasion compared to the wild type stain, which was restored when complemented by plasmid-expression of ipgB1 (Ohya et al., 2005). Membrane ruffle size is also affected, as wild-type strains achieved ruffle size of 60 μm2, an ipgB1 − mutant achieved 16 μm2, and an ipgB1-hyperproducing strain achieved 138 μm2 (Ohya et al., 2005). This indicates that IpgB1 is involved in the production of membrane ruffles in a dose-dependent manner. It was disputed as to whether IpgB1 activated Rac1 directly (Alto et al., 2006) or whether IpgB1 mimicked RhoG for activation of Rac1 indirectly via the ELMO-Dock180 pathway (Handa et al., 2007). However, crystal structures of IpgB1 confirmed that it acts as a GEF, specifically recognizing the β2-3 residues of Cdc42 and Rac1 GTPases to catalyse the GDP-GTP exchange for activation (Huang et al., 2009). Crystal structures and functional studies also confirmed that IpgB2 is a GEF capable of directly binding and activating RhoA (Huang et al., 2009; Klink et al., 2010). Activation of Rac1 and RhoA by IpgB1 and IpgB2 contributes to formation of lamellipodia and actin stress fibers, respectively. In the Sereny test, an ipgB2 − mutant produced the same disease phenotype as the wild-type strain, and a negative result only occurred in an ipgB1 − ipgB2 − mutant, indicating redundancy (Hachani et al., 2007). However, an ipgB1 − mutant alone produced a more severe inflammatory phenotype than the wild-type strain, which was unexpected.
IpaA: Actin Depolymerization
IpaA is involved in regulating actin protrusions at the epithelial membrane and depolymerization of actin filaments in the host cell during Shigella entry. This is postulated to be achieved through its interactions with vinculin, a host protein that links the cytoskeleton to the extracellular matrix and is involved in focal adhesion structures. There are three vinculin binding sites, which are arranged tandemly at the IpaA C-terminus, each of which can bind one vinculin head. Binding of IpaA to the vinculin head induces a conformational change in vinculin, revealing an F-actin binding site in the vinculin tail (Izard et al., 2006). IpaA is important for cell entry, as an ipaA − mutant has a 10-fold decrease in invasion capacity, and requires vinculin to mediate its effects, as vinculin-deficient cells had a similar invasion defect (Tran Van Nhieu et al., 1997). Pelleting assays showed that no actin depolymerization occurred in the presence of IpaA or vinculin alone, with actin principally in the pellet, but when the IpaA:vinculin complex was added, actin was found mostly in the supernatant, with the amount of depolymerization correlating with vinculin concentration (Bourdet-Sicard et al., 1999). The IpaA:vinculin complex has a 3-fold increased affinity for F-actin compared to vinculin alone and acts as a "leaky cap" on the barbed end of the F-actin filaments to prevent addition of further monomers and cause depolymerization (Bourdet-Sicard et al., 1999; Ramarao et al., 2007). IpaA therefore prevents the uncontrolled formation of IpaC-induced microspike structures at the site of bacterial contact, which would repel Shigella from the epithelial cell surface (Tran Van Nhieu et al., 1997).
IpgD: Membrane Ruffles
IpgD plays an important role in the formation of bacterial entry structures on contact with host epithelial cells. An ipgD − mutant induces a less efficient entry structure than the wild type, due to smaller membrane ruffles and a reduction in actin rearrangements (Niebuhr et al., 2000). IpgD functions as an inositol (phosphoinositide) 4-phosphatase, and has sequence motifs similar to mammalian phosphoinositide phosphatases and Salmonella homolog SopB/SigD with similar inositol phosphate phosphatase activity (Niebuhr et al., 2002). Its main substrate in the host cell is phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2/PIP2] which it desphosphorylates to produce phosphatidylinositol 5-monophosphate [PtdIns(5)P/PI5P]. An ipgD − mutant, or IpgD with a cysteine to serine (C438S) substitution in its active site, has no affect on PtdIns(4,5)P2 levels (Niebuhr et al., 2002). The dephosphorylation activity of IpgD on PtdIns(4,5)P2 correlates with a decrease in membrane tether force, as PtdIns(4,5)P2 controls the adhesion force between the plasma membrane of the epithelial cell and the actin cytoskeleton (Niebuhr et al., 2002). Combined with actin cytoskeleton rearrangements mediated by other Shigella effectors, the reduction in membrane tether allows the extension of filopodia and membrane ruffles characteristic of the trigger mechanism seen in Shigella entry into non-phagocytic cells (Figure 1, step 7) (Niebuhr et al., 2002). IpgD inositol-4 phosphatase activity has also been implicated in a positive feedback loop involving ARF6 GTPase (Garza-Mayers et al., 2015), which stimulates actin remodeling and membrane ruffles through Rac1 activation. The production of PtdIns(5)P by IpgD activates phosphoinositide 3-kinase (PI3K), which generates PtdIns(3,4,5)P3. This recruits ARF nucleotide binding site opener (ARNO), a GEF that activates ARF6 GTPase. Active ARF6-GTP promotes actin remodeling through Rac1-dependent pathways, further contributing to membrane ruffles for Shigella entry.
Replication and Spread within Colonic Epithelium
IpaB and IpaC: Lysis of Single Membrane Entry Vacuole?
Due to the pre-requisite for invasion of epithelial cells and time coupling between entry and vacuolar lysis (15 min), disruption of components required for entry can seem to have pleotrophic effects on downstream infection events. This creates intrinsic limitations in studying their role in intracellular pathogenesis (Guichon et al., 2001). Nonetheless, an "entry region" encoding only the T3SS and IpaD, IpaB, and IpaC has been demonstrated to be sufficient for vacuolar lysis (Figure 1, step 8) (Sansonetti et al., 1986; Du et al., 2016). High et al. (1992) used macrophages, which are naturally phagocytic, to overcome the non-invasive ipaB − phenotype, and described how IpaB plays a role in lysis of the phagocytic vacuole. It has been postulated that the insertion of the IpaB-IpaC translocon into the vacuolar membrane is the cause of membrane lysis (High et al., 1992). This would occur through pore formation, which could lead to vacuolar destabilization, or through translocation of unidentified membranolytic effector(s) across the membrane (Page et al., 1999). Senerovic et al. (2012) found that purified IpaB internalized into cells oligomerized in endocytic membranes to form ion channels which affected their integrity. However, the IpaB used was purified recombinantly using a detergent, which naturally lyses membranes, and this effect was not controlled for. Moreover, when within the naturally inserted translocon IpaB is connected to the T3SS via the needle tip, which - along with low osmolarity in the vacuole - would prevent the influx of water into the vacuole to cause lysis. Furthermore, by exploring the functional interchangeability of translocon components from Shigella and Salmonella, which remains in its invasion vacuole, IpaC was shown to be directly involved in lysis of the single membrane vacuole (Osiecki et al., 2001; Du et al., 2016). Yet, any environmental cue for the translocon to switch between possible invasion, translocation, and lysis conformations remains unknown.
IpgD: Lysis of Single Membrane Entry Vacuole?
IpgD may be involved in the modulation of the Shigella-induced entry vacuole by recruiting Rab11 to macropinosomes. An siRNA screen and Rab11-depleted cells showed that the absence of Rab11, a small GTPase involved in endocytic recycling, leads to decelerated Shigella-induced vacuolar rupture. Like the Rab11-depleted cells, vacuolar rupture was delayed in an ipgD − mutant, taking twice as long as for the wild-type strain (Mellouk et al., 2014). Functionally impaired Rab11 and a GDP-locked dominant negative Rab11 showed that it is the absence of Rab11 activity that causes this delay in vacuolar rupture (Weiner et al., 2016). Immunofluorescence staining co-localized Rab11-positive vesicles at the Shigella invasion site and Shigella-containing vacuoles, however this accumulation did not occur if an ipgD − mutant or IpgD lacking its inositol-4 phosphatase activity was present in the vacuole (Mellouk et al., 2014). Using C-FIB/SET, Weiner et al. (2016) then showed that the Rab11-positive vesicles are macropinosomes, which are formed during membrane ruffling induced by Shigella. IpgD is involved in macropinosome formation, through its stimulation of ruffling, and hence in making these organelles available to the Shigella entry vacuole (Weiner et al., 2016). Macropinosomes are required for efficient entry vacuole rupture and have been visualized close to the entry vacuole, making direct contact just prior to vacuolar rupture (Weiner et al., 2016). This suggests that the phosphoinositide phosphatase activity of IpgD is required to regulate Rab11 recruitment to macropinosomes for attachment to the Shigella-containing entry vacuole (Figure 6). How macropinosome attachment, which does not lead to fusion with the vacuole, leads to rapid vacuolar rupture is unknown.
IcsA (VirG): Actin-Based Motility
IcsA is an autotransporter and is composed of three domains: An N-terminal signal sequence, a C-terminal β barrel core which forms a pore in the outer membrane, and a central α-domain which is translocated through the β core membrane pore and present at the Shigella surface (Suzuki et al., 1995). Surface exposed IcsA is sometimes cleaved, however this is not required for IcsA function (Fukuda et al., 1995). The importance of IcsA in Shigella intercellular spread was identified early, as an icsA − mutant was unable to spread within an epithelial monolayer (as measured by plaque formation) and had a negative Sereny test (Bernardini et al., 1989). IcsA-mediated actin based motility is sufficient for membrane protrusion formation (Figure 1, step 10) and entry into neighboring cells (Figure 5). IcsA acts as a mimic of Cdc42 to activate N-WASP, which allows the N-WASP C-terminus to recruit Arp2/3 (Egile et al., 1999; Shibata et al., 2002). This promotes rapid F-actin assembly and filament growth at the N-terminus of N-WASP, providing a propulsive force for Shigella to move through the cell. When icsA is expressed in E. coli, these bacteria are capable of forming membrane protrusions with similar morphology to Shigella-induced protrusions, indicating that no other Shigella factors are required for this process (Monack and Theriot, 2001). Shigella factors, such as IcsP (SopA), are required for correct localization and cleavage of IcsA at the Shigella surface, contributing to efficient motility (Egile et al., 1999). On addition of icsP to icsA-expressing E. coli there was an increase in actin polymerization and increase protrusion frequency. Furthermore, in E. coli LPS O-antigen mutants there was a decrease in formation of actin tails compared to the wild-type. A galU − mutant, which normally encodes a UDP-glucose pyrophosphorylase involved in O-antigen biosynthesis, produces a diffuse circumferential pattern of IcsA on the Shigella surface, which is still capable of polymerizing actin but forms no membrane protrusions (Sandlin et al., 1995). However, it is unknown how the LPS modulates IcsA localization. Therefore, both IcsP and LPS are required for the unipolar localization of IcsA to produce efficient unidirectional movement, which is strongly correlated with frequency of membrane protrusions (Monack and Theriot, 2001).
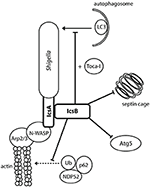
Figure 5. IcsA mediates actin-based motility and IcsB prevents autophagy. IcsA is localized in a unipolar fashion to promote efficient unidirectional intracellular movement parallel to the long axis of Shigella for movement intra- and intercellularly. IcsB masks the Atg5 recognition site on IcsA, recruits Toca-I, and prevents the formation of septin cages.
IcsB: Inhibition of Autophagy
IcsB requires the IpgA chaperone for both its stability and its secretion (Ogawa et al., 2003). An icsB − mutant produced plaques with a smaller diameter than the wild type and a negative result in the Sereny test, suggesting a role for IcsB post-invasion. This role is the prevention of autophagy (Figure 5). IcsB prevents autophagic recognition by masking the Atg5 binding site on IcsA, preventing Atg5 from binding and initiating autophagosome formation. IcsB is also capable of recruiting Toca-I to prevent LC3-mediated phagocytosis (Baxt and Goldberg, 2014). IcsB prevents the formation of septin cages, which in turn may prevent the recruitment of ubiquitin (Ub), p62 and NDP52 (Huang and Brumell, 2014). In an icsB − knockout, autophagic double membranes were visualized around the Shigella bacilli, with asymmetric distribution similar to IcsA placement (Ogawa et al., 2005). IcsB, in conjunction with VirA, has also been implicated in lysis of the double membrane entry vacuole after intercellular spread (Figure 1, step 11). Electron microscopy (EM) 3 h after cell infection visualized icsB − mutants remaining trapped in a double membrane, with several bacteria in one vacuole (Allaoui et al., 1992). Galectin-3 had then been used to show that an icsB − mutant has only a 53% disruption of the double membrane vacuole, compared to 70% disruption mediated by the wild type (Campbell-Valois et al., 2015). However, on closer inspection, we think the EM images (Allaoui et al., 1992, Figure 8B) were misinterpreted, and the double membrane interpreted as the secondary entry vacuole was actually a starting autophagosome wrapping around the icsB − mutant unable to inhibit autophagy. Furthermore, galectin-3 can be used to label endomembranes or autophagic membranes, as it interacts with β-galactose-containing glycoconjugates which are present in both endocytic and secretory compartments (Maejima et al., 2013). Therefore, instead of rupturing the secondary entry vacuole IcsB inhibits autophagy of Shigella.
VirA: Inhibition of Autophagy and Promotion of Golgi Fragmentation
VirA was initially thought to play a role in Shigella invasion, as a virA − mutant had a 5-fold reduced capacity for invasion (Uchiya et al., 1995). This was linked to its apparent cysteine protease activity and capability for microtubule degradation (Yoshida et al., 2002). However, structural analysis showed that VirA lacks the suggested papain-like protease activity for tubulin cleavage, and instead exhibits homology with EspG, an EPEC effector that fragments the Golgi (Germane et al., 2008). VirA belongs to a family of GTPase-Activating Proteins, which share the conserved Rab GTPase catalytic Tre-2/Bub2/Cdc16 domain to mediate Rab1 GTP hydrolysis (Dong et al., 2012). Rab1 GTPase is involved in ER-to-Golgi vesicular transport and is crucial in the formation of autophagosomes (Zoppino et al., 2010). VirA stabilizes Rab1 in the inactive GDP state, thereby directly interfering with autophagy induction and ER-to-Golgi trafficking (Figure 6) (Dong et al., 2012; Huang and Brumell, 2014). A virA − mutant leads to reduced Shigella intercellular persistence, but does not greatly reduce Golgi fragmentation, as IpaJ is more potent in fragmenting the Golgi (Figure 6) (Dong et al., 2012; Burnaevskiy et al., 2013). VirA, similarly to IcsB, has been implicated in the disruption of the secondary vacuole after intercellular spread following membrane protrusion formation (Figure 1, step 10) (Campbell-Valois et al., 2015). However, like for IcsB, this may have been misinterpreted. In our view, the evidence indicates that VirA is involved in lysis of the single membrane entry vacuole (Figure 1, step 8). Indeed, Lysosomal Associated Membrane Protein 2 (LAMP2), a marker for lysosomal fusion with the entry vacuole, has been localized by confocal microscopy to entry vacuoles containing a single virA − mutant and a double icsB − virA − mutant (Campbell-Valois et al., 2015, Figures 2B, 3C) (Dong et al., 2012, Figure S3). A single icsB − mutant is capable of escaping the entry vacuole, visualized by actin comet tail formation (Allaoui et al., 1992, Figure 8A). Therefore, the lack of escape from a single membrane entry vacuole by the double icsB − virA − mutant (Campbell-Valois et al., 2015, Figure 4) can be attributed to loss of VirA function. To confirm this, EM analysis of a single virA − mutant is required. VirA may mediate vacuolar lysis through an indirect mechanism, whereby its inhibition of endosomal trafficking prevents membrane vesicle fusion to the entry vacuole (Figure 6). This could occur through interactions with multiple Rab GTPases, as in vitro assays have indicated that VirA can bind many Rab proteins involved in ER-to-Golgi traffic and recycling (Dong et al., 2012). The vacuole may consequently lyse as it cannot grow to accommodate Shigella replication. Passive lysis of the vacuole has been shown for Salmonella, whereby a sifA − knockout prevents the recruitment of membrane to the entry vacuole. The vacuole cannot sustain Salmonella replication and subsequently lyses, releasing Salmonella into the epithelial cytoplasm (Beuzón et al., 2000).
Figure 6. VirA and IpaJ mediate Golgi fragmentation, which may contribute to entry vacuole disruption. There is a level of semi-redundancy between VirA and IpaJ, as a double virA − ipaJ − knockout is required to completely abolish Shigella-induced Golgi fragmentation. IpaJ is the dominant effector, and targets ARF1-GTP for N-myristoylated cleavage at the exposed di-glycine motif. ARF1 is irreversibly lost from the Golgi membrane, leading to inhibition of Golgi trafficking (Burnaevskiy et al., 2013). VirA is capable of interacting and inactivating many Rab GTPases to mediate Golgi fragmentation, including Rab1 at the ER and Rab33 at the Golgi (Dong et al., 2012). This causes disruption of ER-to-Golgi trafficking, leading to fragmentation of the Golgi. Inactivation of Rab35 at the recycling endosome may prevent recruitment of additional membrane to the Shigella entry vacuole. Replication of Shigella within the entry vacuole without additional membrane may then allow vacuole lysis. IpgD may also contribute to vacuole lysis by recruiting Rab11 to macropinosomes, which have been shown make contact with the entry vacuole to accelerate vacuolar rupture (Mellouk et al., 2014; Weiner et al., 2016).
IpaJ: Golgi Fragmentation
ipaJ is encoded downstream of the ipaBCDA operon and transcribed divergently to it. But the original ipaJ − mutant showed no defect in plaque formation and was Sereny test positive. Therefore, it did not seem to play a crucial role in epithelial invasion or cell-to-cell spread (Buysse et al., 1997). Structural bioinformatics analysis indicated that IpaJ harbored catalytic residues required for peptide bond hydrolysis, and further experiments identified that IpaJ is a cysteine protease which preferentially cleaves N-myristoylated proteins (Burnaevskiy et al., 2013). Although in vitro studies indicated that IpaJ has a large spectrum of N-myristoylated targets (Burnaevskiy et al., 2015, Figure 1D), in vivo it specifically targets ADP-ribosylation factors (ARF), particularly ARF1 (Burnaevskiy et al., 2015, Figure 4D). ARF1 GTPase is localized to the Golgi membrane and the plasma membrane as it plays a role in ER-to Golgi transport, including vesicle formation for cargo transport and maintenance of the Golgi (D'Souza-Schorey and Chavrier, 2006). Removal of the myristoyl group from GTP-active ARF1 by IpaJ causes its irreversible release from the Golgi, inhibiting vesicular trafficking (Figure 6). An ipaJ − mutant has no effect on the ARF1 intracellular pool, but the wild-type strain decreases the amount of ARF1 GTPase bound to the Golgi (Burnaevskiy et al., 2015). Mounier et al. (2012) suggested that IpaB mediates Golgi fragmentation via modulation of the membrane cholesterol concentration, and state that VirA has no obvious effect on disruption. However, the effects of IpaJ were not accounted for, and as the dominant effector in this semi-redundant pair, it is likely that effects attributed to IpaB were actually mediated by IpaJ. The consequences of Golgi disruption by IpaJ are not fully understood but inhibition of STING relocalization from the endoplasmic reticulum (ER) to the intermediate compartment between ER and Golgi (ERGIC) may be one them (Dobbs et al., 2015). STING is a major sensor of cytoplasmic pathogens through detection of DNA and cyclic dinucleotides, where upon it translocates from the ER to ERGIC and activates of the IFN-I pathway.
OspE1 and OspE2: Promotion of Host Cell Adherence to Basement Membrane
OspE1 and OspE2 are 99% identical, which suggests they may have arisen from a gene duplication event (Buchrieser et al., 2000). They are capable of functioning redundantly, however in S. sonnei, ospE1 is a pseudogene (Miura et al., 2006). An ospE2 − knockout caused cellular rounding that was not as a result of apoptosis or necrosis, and wild-type phenotype was restored when ospE2 knockouts were complemented with functioning ospE2 encoded on a plasmid (Miura et al., 2006). OspEs are capable of interacting with integrin-linked kinase (ILK), which is found in the membrane of host cells where it reinforces focal adhesions (Kim et al., 2009). The interaction between OspE and ILK reinforces adhesion contacts between the epithelial cell and the basement membrane (Miura et al., 2006). The OspE-ILK complex interferes with focal adhesion disassembly, reducing focal adhesion kinase phosphorylation and increasing surface β1 integrins (Kim et al., 2009). Tagged OspE was visualized at focal adhesions, however it was diffuse in ILK−/− cells, indicating that ILK is required for OspE membrane localization, and this in turn increases the amount of ILK in the host membrane (Miura et al., 2006). An in vivo inoculation model in the distal colon of guinea pigs showed no Shigella-induced symptoms when a dual ospE knockout was used, however dual complementation of ospE restored the wild-type phenotype, with both inflammation and hemorrhaging (Kim et al., 2009).
IpaB: Cell Cycle Arrest
IpaB has been linked to cell cycle arrest through interactions with Mad2L2, an anaphase promoting complex inhibitor (Iwai et al., 2007). Mad2L2 is involved in promoting entry of epithelial cells into mitosis during G2/M phase by interacting with the Cdh1, an anaphase promoting complex (APC) associated factor involved in preventing mitosis. After mitosis has occurred, Mad2L2 and Cdh1 dissociate and Cdh1 is activated to suppress mitotic cyclins. IpaB interferes with Mad2L2-Cdh1 binding, causing Cdh1 to be constitutively activated (Iwai et al., 2007). Permanent mitotic cyclin suppression by Cdh1 prevents epithelial turnover during Shigella infection, promoting more efficient bacterial replication by keeping the cells better attached to the adjacent cells and to the lamina. Cell cycle arrest thereby prevents epithelial cell turnover and allows Shigella to establish a better niche for replication. Interactions with Mad2L2 allow IpaB to be translocated into the epithelial nucleus (Iwai et al., 2007). IpaB binds Mad2L2, and this occurs at the same location on IpaB where the IpgC chaperone binds. Introduction of a single amino acid substitution conferring weaker binding of IpaB to Mad2L2 leads to a reduction in colonization of rabbit ileal loops suggesting that IpaB and its interaction with Mad2L2 contributes to more efficient Shigella colonization of the epithelium (Iwai et al., 2007, Figure 6B). However, the point mutation could instead have pleotropic effects on IpaB function (see IpaB and IpaC: Lysis of single membrane entry vacuole?) rather than directly affecting Mad2L2 binding.
Lysis of the Double Membrane Vacuole
Vps and VacJ: Proposed ABC Transporter
The vpsABC operon is found on the Shigella chromosome, and consists of VpsA, a possible ATP-Binding Cassette (ABC) transporter protein, and VspB and VspC, proposed transmembrane proteins. Both vpsC − and vspA − knockouts had a defect in plaque formation but were similar to wild-type strains in their capability to invade, indicating that they play a role in intercellular spread (Hong et al., 1998). VacJ is also encoded on the chromosome, and a vacJ − knockout is incapable of escaping into the recipient cell cytoplasm, suggesting that VacJ also plays a role in intercellular spread (Suzuki et al., 1994). Carpenter et al. (2014) describe a Vps/VacJ ABC transporter, which maintains asymmetry of lipids in the outer membrane and in the context of Shigella infection is required for lysis of the double membrane vacuole. Transformation of vps/vacJ knockouts with a plasmid expressing pldA, a phospholipase in other Gram negative bacteria, was able to restore the maintenance of outer membrane lipid asymmetry but was unable to lyse the double membrane vacuole, indicating that these two functions of the proposed Vps/VacJ ABC transporter are separate (Carpenter et al., 2014). Another substrate may therefore be transported across the membrane to induce vacuolar lysis, however this is yet to be discovered.
IpaB and IpaC: Translocon Formation and Lysis of the Double Membrane Vacuole
Studying intracellular roles of IpaB and IpaC is difficult as their non-invasive mutants have pleotropic effects. However, Page et al. (1999) overcame this issue using a recombinant plasmid with IPTG-inducible lac promoter to regulate expression of IpaB and IpaC. After initial entry, IPTG was removed from the medium, producing ipaB − and ipaC − phenotypes which have a 3-fold decrease in plaque diameter compared to the wild-type. The inducible ipaB − or ipaC − mutants both exhibited a defect in lysis of the double membrane vacuole (Figure 1, step 11), with abolished membrane contact formation (Campbell-Valois et al., 2014). Several bacteria were visualized within such vacuoles, indicating that enough time was spent in the vacuole for replication to occur (Page et al., 1999). IpaB is located at the needle tip along with IpaD, from where it senses the host cell membrane (Murillo et al., 2016). Therefore, its interactions with the inner surface of the plasma membrane during membrane protrusion formation may induce the on-off regulation of the T3SS in the epithelial cell cytoplasm (Campbell-Valois et al., 2014) and fresh translocon insertion and/or translocation of unidentified effector(s) involved in lysis of the first or second membrane of the double membrane vacuole.
Modulation of Innate Immune System
We cover here effectors directly involved in suppressing the innate immune response in epithelial cells and/or macrophages, rather than events up-stream of it discussed above, such as autophagy.
OspC3: Inhibits Caspase-4-Mediated Inflammatory Cell Death
OspC3 is part of the ospC gene family. An ospC3 − mutant has an increased inflammatory cell death when compared to wild-type Shigella and ospC1 − /ospC2 − mutants (Kobayashi et al., 2013). This indicates that OspC3 plays a role in the down-regulation of acute inflammatory cell death, and suggests a lack of redundancy in the ospC family as OspC1 has pro-inflammatory effects. Inflammatory cell death was not abolished with a cytochrome c or caspase-3/caspase-7 inhibitor. However, a caspase-1/caspase-4/caspase-5 inhibitor did reduce cytotoxicity and their activity increased during ospC3 − infection, suggesting that OspC3 mediates its activity via one of these caspase pathways (Kobayashi et al., 2013). Tagged OspC3 bound to the caspase-4 p19 subunit in a pull-down assay, and in-frame deletions showed that the terminal 190–484 residues of OspC3 were involved in p19 binding, specifically at a conserved consensus sequence, X1-Y-X2-D-X3 (Kobayashi et al., 2013). Substitution of all five residues with alanine, in addition to substitution of the conserved 450–478 residues in the C-terminal ankyrin region, impaired OspC3 binding to p19 and increased epithelial cytotoxicity (Kobayashi et al., 2013). Incubation of the p19 subunit with increasing concentrations of OspC3 correlated with increasingly impaired p19–p10 binding (Kobayashi et al., 2013). The biochemical function of OspC3 may therefore be interacting with p19 to inhibit caspase-4 activation and prevent inflammatory cell death (Figure 7).
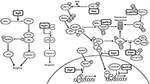
Figure 7. Shigella modulates the innate immune response in epithelial cells. NFκB can be activated via many pathways during Shigella infection, including genotoxic stress during invasion and recognition of the lipopolysaccharide on the bacterial surface. This may explain the redundancy of Shigella effectors, many of which have the ability to interfere with these pathways, leading to efficient inhibition of NFκB. IpgD increases levels of PI5P, which activates PI3K and Akt and inhibits p53 and caspase 3. OspC3 sequesters the p19 pre-cursor of caspase 4, which prevents its heterodimerization with p10 and inhibits capase 4 activation. OspI deamidates Ubc13, which inhibits ubiquitination of TRAF6 and prevents activation of the TRAF6-NFκB pathway. IpaH0722 ubiquitylates TRAF2, leading to its proteasomal degradation and preventing recruitment of IKK and activation of NFκB. Polyubiquitination of NEMO/IKKγ is dependent on IpaH9.8 binding to A20 binding inhibitor of NFκB (ABIN-1), leading to proteasomal degradation of NEMO/IKKγ, activation of IκBα, and inhibition of NFκB. IpaH9.8 also localizes to the nucleus where it catalyses ubiquitination of U2AF35, leading to its degradation and consequently reduced il-8 expression and decreased neutrophil recruitment. By an unknown mechanism, OspG prevents SCFβ−TrCP from ubiquitinating IκBα, preventing degradation of IκBα and activation of NFκB. OspZ blocks the nuclear translocation of p65, a subunit of NFκB, thereby inhibiting activation of NFκB. Threonine desphosphorylation of MAPKs by OspF leads to inactivation of ERK1/2 and p38 MAPK pathways and inhibition of histone H3 phosphorylation (H3pS10). This produces an inaccessible chromatin conformation at the gene promoters of inflammatory cytokines and chemokines, meaning NFκB cannot activate their transcription.
OspF: Inactivates MAPKs Which Prevents Phosphorylation of Histone H3
OspF was first described as a dual specific phosphatase (Arbibe et al., 2007), desphosphorylating threonine and tyrosine residues in the MAPK signaling pathway (Figure 7). However, it was determined by Li et al. (2007) through tandem mass spectrometry that OspF instead displays phosphothreonine lyase activity in that it irreversibly desphosphorylates threonine, but not tyrosine, through beta elimination (Zhu et al., 2007). An ospF − mutant has increased PMN recruitment and severity of epithelial destruction in the rabbit ileal loop model when compared to the wild-type strain, indicating a role in down-regulating the immune response to Shigella infection (Arbibe et al., 2007). Phosphothreonine lyase activity is also seen in SpvC, a Salmonella homolog with 63% amino acid identity with OspF. Antibodies against phospho-amino acids confirmed specific removal of phosphate from threonine which inactivates MAPKs (Mazurkiewicz et al., 2008). OspF has also been attributed pro-inflammatory roles (Zurawski et al., 2006; Reiterer et al., 2011). The identification of accurate in vivo substrates may explain the apparent pro- and anti-inflammatory roles mediated by OspF.
OspG: Inhibits NFκB Activation
OspG plays a role in dampening the host immune response, shown by an ospG − mutant exhibiting increased inflammation and destruction of the mucosa in comparison to wild-type Shigella in the rabbit ileal loop model (Kim et al., 2005). OspG has a minimal kinase domain, and its kinase activity requires binding of an E2 ubiquitin conjugating enzyme in conjunction with ubiquitin (Pruneda et al., 2014). Binding of UbcH7~Ub stabilizes OspG and confers an active kinase conformation, increasing kinase activity 20-fold (Grishin et al., 2014). OspG is also capable of binding UbcH5b~Ub, which is a component of the E3 ligating enzyme SCFβ−TrCP (Kim et al., 2005). An ospG − mutant exhibits IκBα degradation 20 min post-invasion, whereas in wild-type Shigella this degradation occurs after 60 min (Kim et al., 2005). The exact mechanism of how OspG prevents SCFβ−TrCP from ubiquitinating phospho-IκBα is unknown, but the OspG kinase activity is postulated to be involved in the attenuation of NFκB activation (Figure 7) (Zhou et al., 2013).
OspI: Inhibits NFκB Activation
OspI functions as a glutamine deamidase, and has been shown to interfere with NFκB activation via the TNF-receptor-associated-factor (TRAF) 6 pathway (Sanada et al., 2012). An ospI − mutant, when compared to wild-type Shigella, leads to increased levels of cytokine mRNA transcripts, increased phosphorylation of IκBα and a 4-fold increase in nuclear translocation of the p65 subunit of NFκB (Sanada et al., 2012). All of these lead to increased NFκB activation and consequently an increase in the host inflammatory response. OspI has a cysteine-histidine-aspartic acid catalytic triad which is crucial for deamidation, as activity was abolished by a cysteine-to-serine substitution (Sanada et al., 2012). A substrate of OspI is UBC13, an E2 ubiquitin conjugating enzyme required for TRAF6-induced NFκB activation, shown by binding of OspI to His-UBC13 during pull-down assays (Nishide et al., 2013). Hydrophobic interactions are important for this binding, and a crystal structure shows a glutamine residue at position 100 on UBC13 is positioned in the OspI active site (Nishide et al., 2013). OspI specifically deamidates Gln100, converting it to glutamic acid and abolishing the E2 ubiquitin conjugating function of UBC13 to prevent activation of the TRAF6-NFκB pathway (Figure 7) (Sanada et al., 2012).
OspZ: Inhibits NFκB Activation
As previously described, OspZ has a pro-inflammatory role in some S. flexneri species. In the remaining Shigella species the full length OspZ has an anti-inflammatory role, similar to that of its NleE homolog in EPEC (Newton et al., 2010). Both NleE and OspZ have been shown to block the nuclear translocation of p65, a subunit of NFκB, in response to TNFα and IL-1β (Figure 7). This leads to a reduction in transcription of pro-inflammatory cytokine genes, such as il-8, thereby reducing inflammation during Shigella infection. OspZ and NleE are also capable of inhibiting IκB degradation, further suppressing NFκB activity. Newton et al. (2010) determined that the crucial region for the anti-inflammatory effect of OspZ and NleE was between the amino acids 208–214, and has the sequence IDSYMK. Deletion of this region or single amino acid substitutions for alanine in NleE led to an increase in NFκB-dependent transcription (Newton et al., 2010, Figure 6D). NFκB activity was not abolished, indicating that this region is a binding site rather than an enzyme active site. The precise mechanism of how OspZ inhibits IκB degradation is unknown. However, Nadler et al. (2010) propose that NleE inhibits IKKβ, which is normally responsible for IκB phosphorylation and degradation in response to pro-inflammatory stimuli. OspZ may therefore work by a similar mechanism to prevent NFκB activation.
IpaH9.8: Inhibits NFκB Response
IpaH9.8 is a member of the ipaH gene family, and one of four found on the virulence plasmid, which includes ipaH1.4, ipaH2.5, ipaH4.5, and ipaH9.8. IpaH proteins are characterized by an N-terminal leucine-rich repeat (LRR) region and a highly conserved C-terminal region (CTR) which contains a cysteine residue (Suzuki et al., 2014a). The LRR motif is thought to play a role in protein-protein interactions, such as cell adhesion and signaling, while the cysteine residue in the CTR is required for enzyme 3 (E3) ubiquitin ligase activity. An ipaH9.8 − mutant has an increased inflammatory phenotype in comparison to wild-type Shigella in the murine lung model, indicating that IpaH9.8 has a role in attenuating inflammation during Shigella infection (Okuda et al., 2005). This is achieved through E3 ubiquitin ligase activity via the CTR of IpaH9.8 (Rohde et al., 2007). The substrates of IpaH9.8 include U2AF35and NEMO/IKKγ (Okuda et al., 2005; Ashida et al., 2010). Pull down assays confirmed IpaH9.8 and U2AF35 binding, which specifically occurs at the C-terminus of IpaH9.8 and 107–197 residues on U2AF35 (Okuda et al., 2005). Binding of IpaH9.8 to NEMO/IKKγ requires an ABIN-1 (A20 Binding Inhibitor) adaptor, as ABIN-1 knockdown leads to lack of IpaH9.8-induced effect on NEMO levels (Ashida et al., 2010). Ubiquitination of both U2AF35 and NEMO/IKKγ mediated by IpaH9.8 leads to their degradation in a proteasome-dependent manner (Figure 7) (Ashida et al., 2010; Perrett et al., 2011). Seyedarabi et al. (2010) describe how IpaH9.8 domain swapping occurs in response to host cell damage and this leads to dimerization and inactivation of its E3 ubiquitin ligase activity. Shigella may therefore sense the host cell conditions to maintain a suitable environment for its continued proliferation and survival.
IpaH0722: Inhibits NFκB Activation
IpaH0722 is encoded on the Shigella chromosome. In an ipaH-null mutant, whereby all seven of the chromosomal ipaH family genes were deleted, there was an increase in the severity of inflammation in the murine lung model in comparison to wild-type Shigella infection (Ashida et al., 2007). When individual ipaH knockouts were examined, it was found that IpaH0722 plays a role in dampening the inflammatory response, as an ipaH0722 − knockout had increased levels of IκBα degradation leading to NFκB activation (Ashida et al., 2013). IpaH0722 is also an E3 ubiquitin ligase, with the conserved CTR and crucial cysteine residue that is found in other IpaH proteins. A cysteine-to-alanine substitution increased NFκB activation, indicating that the E3 ubiquitin ligase activity is crucial for downregulation of NFκB (Ashida et al., 2013). Pull down assays showed that IpaH0722 could bind to TRAF2, however a CTR-truncation was unable to interact with TRAF2, further indicating the importance of IpaH0722 ubiquitin ligase activity (Ashida et al., 2013). IpaH0722 causes increased TRAF2 degradation leading to a reduction in NFκB activity, thereby dampening the host inflammatory response (Figure 7).
IpgD: Activates Akt/PI3K Signaling Pathway
IpgD-mediated increase of PtdIns(5)P has been shown to induce Akt phosphorylation through activation of phosphatidylinositol 3-kinase (PI3K) (Mayo and Donner, 2001). An ipgD − knockout has abolished Akt phosphorylation, which also occurs if PtdIns(5)P is sequestered or phosphorylated to PtdIns(4,5)P2 (Pendaries et al., 2006). Reduction in Akt phosphorylation was correlated with an increase in apoptosis and decreased phosphorylation of Mdm2, the negative regulator of p53, by the Akt serine-threonine kinase (Mayo and Donner, 2001). Bergounioux et al. (2012) showed that an ipgD − mutant had a reduction in early phase Mdm2 phosphorylation, causing a delay in p53 degradation and increased apoptotic phenotype. The Salmonella homolog SopB and SigD also have pro-survival functions through interactions with Akt and activation of the PI3K-Akt survival pathway (Steele-Mortimer et al., 2000; Knodler et al., 2005).
Macrophage Vacuolar Rupture and Pyroptosis
IpaC: Rupture of Phagocytic Vacuole
IpaC interacts with IpaB at the tip of the T3SS needle to form a translocon that inserts into lipid membranes (Blocker et al., 1999). An ipaC − mutant has no haemolytic activity and is unable to escape the phagocytic vacuole (Bârzu et al., 1997). Given the role determined for IpaC in lysis of the invasion vacuole in epithelial cells (Osiecki et al., 2001; Du et al., 2016), it is very likely that IpaC also plays a role in this process in macrophages (Figure 1, step 3). Salmonella-induced vacuoles in macrophages have an acidic pH, and it was proposed that acidification may also be the cue to change IpaC function from cell entry to membrane lysis (De Geyter et al., 1997). If acidification is not the cue for vacuolar lysis, blocking acidification of endosomes using Bafilomycin-A1 should not prevent Shigella from exiting the vacuole.
IpaB: Promotes Macrophage Pyroptosis
IpaB's hydrophobic region (310–430 amino acid residues) has 65% sequence identity to Salmonella invasive protein B (SipB), and is involved in invasion of the epithelium via translocon formation, phagosome escape, and induction of macrophage pyroptosis (Guichon et al., 2001). The binding site for the pro-pyroptotic and pro-inflammatory caspase-1, also known as interleukin-1β converting enzyme (ICE), is located at residues 311–401 within the hydrophobic region (Guichon et al., 2001). IpaB is localized at the bacterial surface and in discrete aggregates in the macrophage cytoplasm, which suggests that IpaB interacts with caspase-1 after vacuolar lysis rather than being injected into the cytoplasm from the vacuole (Thirumalai et al., 1997). The IpaB-ICE complex cleaves the precursors of pro-inflammatory cytokines IL-1β and IL-18 to produce mature IL-1β and IL-18, which are released in parallel to the induced pyroptosis (Figure 1, steps 4, 6). IpaB may also promote macrophage pyroptosis by allowing delivery of the T3SS the needle and rod proteins, MxiI and MxiH, into the cytosol. These bind the NAIP family of inflammasome receptors that trigger activation of caspase-1 (Yang et al., 2013; Suzuki et al., 2014b). Recent work also suggests that IpaD promotes macrophage apoptosis independent of caspase-1 but via host caspases accompanied by mitochondrial disruption (Arizmendi et al., 2016).
IpaH7.8: Promotes Macrophage Pyroptosis
IpaH7.8 is part of the ipaH gene family found on the Shigella virulence plasmid. It was suggested that IpaH7.8 had a role in vacuolar lysis as an ipaH7.8 − mutant show reduced from escape the phagocytic vacuole (Fernandez-Prada et al., 2000). However, how this mutant strain (PWR700) was made is unclear, and its complementation was poor. Paetzold et al. (2007) then described how an ipaH7.8 − knockout was able to escape the phagosome. Therefore, IpaH7.8 has no role in vacuolar escape. Instead, the IpaH7.8 E3 ubiquitin ligase targets glomulin, an inhibitor of inflammasome activation, for ubiquitination leading to glomulin degradation. Macrophage-specific cell death (Figure 1, step 6) is then triggered through activated inflammasomes (Suzuki et al., 2014a).
Modulation of Adaptive Immune System
LPS—Serotype Conversion and Thymus-Independent T-cell Activation
The acquisition of the SHI-O PAI means that Shigella is capable of modifying its LPS (Figure 8). As humoral immunity is serotype specific, and protection against re-infection is often unsuccessful because of the variety of Shigella serotypes. Furthermore, O-antigen of LPS is a carbohydrate, thymus-independent type 1 (TI-1) antigen, which activates B-cells in the absence of helper T-cells (Murphy, 2012). The lack of helper T-cell involvement means that these activated B-cells cannot undergo class switching or develop a memory B-cell response to protect against re-infection. At a low concentration of TI-1 antigens, such as when LPS molecules are released from damaged bacteria, the naïve B-cells are activated due to specific binding of their B-cell receptors to the antigen (i.e., O-antigen of LPS). This induces the production of O-antigen-specific antibodies, which are protective against Shigella infection, however they are not long-lasting and are overcome by serotype conversion. A high concentration of TI-1 antigens, like the O-antigen on LPS at the bacterial surface, leads to non-specific polyclonal activation of B cells and the production of non-specific and hence likely non-protective antibodies (Murphy, 2012). Thus, O-antigen behaves as an "immunological decoy" at more than one level, making it a particularly poor choice as a vaccine antigen.
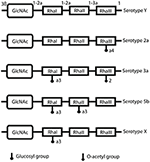
Figure 8. S. flexneri serotype composition and conversion. LPS O-antigen modifications include glucosylation (the addition of glucosyl groups), mediated by the gtr operon and the addition of O-acetyl groups, achieved by the O-acetyltransferase protein encoded by the oac gene. This is the basis of serotype conversion, as Shigella begins with serotype Y and the basic O-antigen structure, and further modification produces different serotypes (Allison and Verma, 2000).
IpgD: Interferes with T-Lymphocyte Migration
IpgD is capable of interfering with T-cell migration by de-phosphorylating PtdIns(4,5)P2 (Figure 9). Wild-type Shigella causes a 50% decrease in T-cell migration toward a chemokine, such as CXCL12 (Konradt et al., 2011). Cells transfected with IpgD-GFP and incubated with CXCL12 had no ERM protein localization at the pole, preventing T-cell polarization and migration (Konradt et al., 2011). Mean velocity of T-cells was measured in vivo, with uninfected T-cells exhibiting 9 and 4 μm/min for wild-type Shigella-infected T-cells (Salgado-Pabón et al., 2013). This indicates that Shigella is capable of affecting T-cells in the context of infection, and that this ability is dependent on IpgD. Shigella may prevent T-cell migration and recruitment to areas of infection as they could be primed by CD1 antigen-presenting cells, which are involved in the presentation of lipid antigens, including LPS (Murphy, 2012). This could induce a specific antigen-response as priming of T-cells leads to stimulation of B-cells for isotype switching and immunological memory, therefore it is advantageous to prevent T-cell involvement in the adaptive immune response. Reducing migration of CD8+ cytotoxic T-cell may also slow clearance of infected cells from the epithelium.
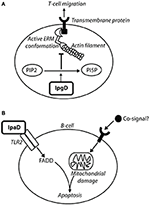
Figure 9. IpgD inhibits T-cell migration and IpaD induces B-cell apoptosis. (A) The PIP2 concentration at the plasma membrane is responsible for the dynamic interchange between active and inactive conformations of ezrin, radixin, and moesin (ERM) proteins (Konradt et al., 2011). ERM proteins are involved in cell-cortex organization of T-cells, and their active conformations localize at the membrane in response to chemokine stimulation to allow T-cell migration toward chemoattractants (Konradt et al., 2011). In the subcapsular sinus of the lymph node (Salgado-Pabón et al., 2013) Shigella can invade T-cells or inject IpgD via the T3SS into T-cells to reduce PtdIns(4,5)P2 concentration. This prevents cell polarisation induced by active ERM proteins and T-cell migration to sites of infection (Konradt et al., 2011). (B) Bacterial co-signals increase pro-apoptotic proteins, induce loss of mitochondrial membrane potential (MMP), and also upregulate tlr2 mRNA, leading to increased TLR2 expression on the surface of the cell. IpaD signaling via the TLR2-1 heterodimer leads to up-regulation of FAS-associated death domain (FADD) protein, which ultimately induces B-cell apoptosis (Nothelfer et al., 2014).
IpaD: Promotes B-Lymphocyte Apoptosis
To prevent an antibody response to the LPS, specific or non-specific, and production of immunological memory, Shigella also induces apoptosis in B-cells. This is mediated by IpaD in a manner independent of Shigella invasion and effector injection (Nothelfer et al., 2014). Incubation of B-cells with IpaD alone does not induce cell death, and it was deduced that bacterial co-signals work in conjunction with IpaD to mediate B-cell apoptosis (Figure 9) (Nothelfer et al., 2014). When anti-IpaD antibodies are applied to rectal biopsies of shigellosis patients, they are visualized within isolated lymphoid follicles and are contacting B-cells, suggesting that IpaD-induced B-cell apoptosis occurs in vivo (Nothelfer et al., 2014).
Discussion
How Do the Virulence Factors Work Together?
Epithelial Barrier Destabilization and Fluid Secretion Causes Severe Intestinal Inflammation and Diarrhoea
Once ingested, S. flexneri 2a delivers ShET1 in the jejunum to elicit fluid secretion (Fasano et al., 1997). Other Shigella species that harbor ShET2 require activation of the T3SS for its secretion and this most likely occurs later after contact with the colonic epithelium (Farfán et al., 2011). Pic and SigA, also harbored by S. flexneri 2a species, act within the lumen of the colon, on the apical side of the epithelium. Pic degrades the thick mucus layer by its mucinolytic activity to give Shigella better access to the epithelium (Gutierrez-Jimenez et al., 2008). SigA mediates enterotoxic activity similar to ShET1 (Al-Hasani et al., 2000) and may have cytopathic effects to initially destabilize the epithelial barrier (Al-Hasani et al., 2009). SepA causes fluid secretion in S. flexneri 5a (Benjelloun-Touimi et al., 1995). Shigella traverses the epithelium via M cells, and reaches the basolateral surface of the epithelium. Here the T3SS is activated, and Shigella can produce ShET2 to induce further fluid secretion (Nataro et al., 1995). Other T3SS effectors, including OspB (Zurawski et al., 2009; Ambrosi et al., 2014), OspC1 (Zurawski et al., 2006), and OspZ (Zurawski et al., 2008) (in S. flexneri) are secreted to phosphorylate and activate MAPK pathways. Consequently, increased apical secretion of the chemoattractant IL-8 causes PMN leukocytes to migrate across the epithelium in a basolateral to apical direction. This destabilizes the epithelial barrier, and the remaining bacteria at the apical surface can access the basolateral surface. Shigella that are phagocytosed by macrophages mediate pyroptosis via IpaB, T3SS components and probably LPS, which bind and activates caspase-1 (Thirumalai et al., 1997), and IpaH7.8 E3 ubiquitin ligase, which targets glomulin, an inflammasome inhibitor, for degradation (Suzuki et al., 2014a). Pyroptosis leads to release of pro-inflammatory cytokines IL-1β and IL-18, which further recruit PMN leukocytes and increase inflammation. All of these virulence determinants and effectors are therefore involved in the characteristic inflammation seen in shigellosis, including the watery diarrhea seen in the early stages. In S. flexneri 2a infection, mucus and blood may also be present due to the mucin secretagogue activity of Pic, and the cytopathic effects of SigA. Shiga toxin, found only in S. dysenteriae, may cause severe bloody dysentery by damaging the vascular endothelium of the colon (Fontaine et al., 1988).
Adhesion and Entry to Epithelium Causes an Epithelial Genotoxic Stress Response
At the basolateral surface, glucosylation of the LPS is induced by an unknown trigger, and allows the T3SS to access the epithelium for activation (West et al., 2005). IpaB at the tip of the T3SS needle complex may bind to CD44 lipid microdomains to maintain contact of the T3SS with the epithelial cell (Skoudy et al., 2000), and IcsA increases polar adhesion (Brotcke Zumsteg et al., 2014). IpaB is inserted into the epithelial membrane, activating IpaD and leading to recruitment of IpaC to the host membrane to form the translocon pore (Blocker et al., 1999). IpaD activation also signals for the T3SS to facilitate early effector translocation through the T3SS needle and into the epithelial cell (Roehrich-Doenitz et al., 2013). IpaC, as one of the immediate effectors accessing the epithelial cell, mediates actin polymerization, either independently or indirectly via Cdc42 and Rac1 (Tran Van Nhieu et al., 1999; Mounier et al., 2009). This initiates membrane ruffles and entry foci, further promoted by IpgB1 and IpgB2, which remodel actin and the host cytoskeleton via their GEF activity (Huang et al., 2009). IpaA, in conjunction with host proteins, facilitates actin depolymerization, interfering with focal adhesions and preventing uncontrolled actin polymerization by IpaC (Tran Van Nhieu et al., 1997; Bourdet-Sicard et al., 1999). IpgD converts PtdIns(4,5)P2 to PtdIns(5)P, which reduces the membrane tether force and further stimulates membrane ruffle formation (Niebuhr et al., 2002). Together these effectors mediate the trigger mechanism for Shigella uptake into non-phagocytic epithelial cells. Upon enclosure in the primary entry vacuole, Shigella mediates membrane lysis. This involves primarily IpaB and/or IpaC in the translocon pore (Osiecki et al., 2001; Du et al., 2016), and as accessory processes IpgD-mediated recruitment of Rab11 to macropinosomes which make contact with the entry vacuole (Weiner et al., 2016), and/or VirA preventing growth of the vacuole membrane (Mellouk et al., 2014).
Intercellular Spread and Immune Evasion Causes Erratic Epithelial Destruction and Prevention of Immunological Memory
Once inside the epithelial cytoplasm, Shigella uses VirA and IpaJ to inactivate mainly Rab1 and ARF6, respectively. This halts ER-to-Golgi traffic, preventing autophagic membrane formation and inducing Golgi fragmentation (Dong et al., 2012; Burnaevskiy et al., 2013, 2015). Shigella further prevents autophagy using IcsB to mask the Atg5 binding site on IcsA (Ogawa et al., 2005). Unipolar localization of IcsA at the Shigella old pole, possibly achieved by LPS and IcsP, is crucial for efficient unidirectional movement (Monack and Theriot, 2001) and allows Shigella to move through the epithelial cytoplasm until it makes contact with the inner surface of the plasma membrane. It then protrudes into the adjacent epithelial cell, forming a secondary entry vacuole consisting of a double membrane. This is subsequently lysed by an unknown mechanism to allow access into the adjacent epithelial cell cytoplasm. Remaining within the epithelial layer is important for renewing the Shigella replication niche and also evading immune detection. Evasion of the immune response is achieved by T3SS effectors that inhibit activation pathways of NFκB, including OspG (Kim et al., 2005), OspI (Sanada et al., 2012), OspZ (Newton et al., 2010), IpaH9.8 (Ashida et al., 2010), and IpaH0722 (Ashida et al., 2013). Degradation of the pro-apoptotic factor p53 is achieved by IpgD (Mayo and Donner, 2001). Inhibition of the innate immune response also prevents development of an adaptive immune response. The movement of Shigella through the epithelium and the subsequent necrotic epithelial cell death is the cause of the colonic destruction and abdominal pain in shigellosis patients, contributing to prevention of fluid absorption and dysentery containing blood.
Virulence Determinants in Disease-Causing Species
S. flexneri has a relatively stable genome, and acquired the virulence plasmid early in its evolution. S. flexneri is capable of persisting in water for several months, similarly to Vibrio cholera (Faruque et al., 2002). This may explain its epidemiological prevalence, as access to human hosts for months at a time could facilitate the endemics seen in countries with poor water sanitation. Speculatively, it may cause the most disease as S. flexneri 2a harbors the SHI-1 PAI, which encodes Pic, SigA, and ShET1. Pic may confer an advantage for scavenging nutrients, therefore other strains may have a metabolic disadvantage when compared to S. flexneri (Henderson et al., 1999; Harrington et al., 2009). The importance of Pic and ShET1 in pathogenesis has also been highlighted by Kotloff et al. (2004). The deletion of pic and set1AB, in addition to ospD3, in a guanine autotrophic (guaAB −) background produced an increasingly attenuated vaccine, with none of the 14 volunteers developing diarrhea (Kotloff et al., 2004). The truncated OspZ found in S. flexneri has a pro-inflammatory role, compared to its anti-inflammatory in the remaining sub-species, which may confer an inflammatory advantage for initial establishment of infection (Zurawski et al., 2008).
S. sonnei causes endemics in industrialized countries. Its emergence was defined by its acquisition of the pINVB plasmid, which harbors genes for the Plesiomonas shigelloides-related serotype 17 O-antigen (Shepherd et al., 2000). P. shigelloides, similarly to S. flexneri, can contaminate and persist in water sources. Infection with P. shigelloides from contaminated water may protect human hosts from established S. sonnei infection due to immunological memory and cross-reactive antibodies to the identical O-antigen (Sack et al., 1994). This hypothesis could also explain why an improvement in water sanitation causes a decrease in S. flexneri infection but an increase in S. sonnei infections, as there is no previous infection with P. shigelloides from contaminated water to induce a protective immunological memory response. Direct transmission of S. sonnei in schools and care settings is also capable of maintaining endemics in communities, without the requirement of an environmental reservoir.
S. dysenteriae infection is the most severe, which has been linked to the effects of the Shiga toxin harbored by S. dysenteriae type 1. There is no known natural reservoir for S. dysenteriae, and it causes sporadic epidemics linked to poor hygiene and overcrowding. The Shiga toxin does not play a role in the intracellular infection, but can stimulate the recruitment of PMN leukocytes and damages the colonic vascular endothelium, leading to the characteristic blood-containing dysentery (Fontaine et al., 1988). It has been suggested that, like Salmonella enterica serovar Typhi, S. dysenteriae can be maintained and transmitted within a community by an asymptomatic carrier. Long term carriers with attenuated symptoms have been previously reported (Levine et al., 1973; Clements et al., 1988), and this may explain the epidemiology of S. dysenteriae epidemics, which disappear to then reappear years later, and are also transferred intercontinentally (Rohmer et al., 2014).
S. boydii causes the least burden of disease worldwide. It represents 1–2% of Shigella isolated, and is mostly confined to the Indian subcontinent. In 2000, S. boydii serotype 20 was discovered. Its transmission was linked to travel in Mexico (Kalluir et al., 2004) and it was the most frequent agent of S. boydii infection in Canada (Woodward et al., 2005). However, S. boydii still only caused 1% of Shigella infections in the United States compared to the 77% of infections caused by S. sonnei (Kalluir et al., 2004). Due to the diversity of S. boydii serotypes, and its lack of clinical relevance there is little known about its virulence determinants and why it causes less disease than S. flexneri, S. sonnei, and S. dysenteriae.
Effector Interplay
Multiple Effectors for One Target Pathway
Effector-Triggered Immune Pathology (ETIP) is an advanced model of both the Guard Hypothesis and Effector-Triggered Immunity (ETI). If the host is "resistant" and encodes the guardee protein, it is capable of recognizing the damage mediated by Shigella effectors and this induces an efficient innate immune response. Shigella can use this to its advantage, increasing intestinal inflammation which is required for infection establishment and transmission by diarrhea (Stuart et al., 2013). However, this inflammation eventually leads to the clearance of infection, therefore Shigella harbors a large repertoire of anti-inflammatory mechanisms for down-regulation of ETIP. The host is not able to "guard" all the steps/signaling pathways involved in NFκB activation, therefore multiple effectors with different biochemical functions are capable of efficiently interfering with these pathways and dampening the host immune response. Similarly, VirA and IpaJ act to fragment the Golgi via exertion of differing biochemical activities on different small GTP binding proteins involved in its maintenance.
More Than One Effector with a Similar Biochemical Function
IpgB1 and IpgB2 are similar in their WxxxE motif and GEF activity, but they have non-overlapping substrates. Only a dual ipgB1 − ipgB2 − knockout had a negative Sereny test, indicating that they also have similar but non-overlapping functions (Hachani et al., 2007). Lack of redundancy was also confirmed, as an ipgB2 − knockout has the same phenotype as the wild-type strain, however the ipgB1 − knockout had an increase in inflammation compared to the wild-type strain. This may link to the guard hypothesis, whereby IpgB1 prevents detection of IpgB2 by the host guardee proteins, perhaps via its own non-overlapping enzyme activity, to prevent an excessive inflammatory response.
One Effector with Multiple Functions?
An effector characterized with multiple roles in pathogensis is normally due to a single function which mediates several effects. For example, the only characterized activity of IpgD is that it dephosphorylates phosphoinositides, decreasing levels of PtdIns(4,5)P2 and increasing levels of PtdIns(5)P. Changes in the intracellular concentrations of these phosphoinositides then produces effects directly, such as membrane ruffles (Niebuhr et al., 2002), or indirectly, such as vacuolar lysis (Mellouk et al., 2014), activation of Akt/PI3K signaling pathway (Pendaries et al., 2006), and interference with T-lymphocyte migration (Konradt et al., 2011). Therefore, IpgD has one biochemical activity, which mediates multiple effects on the host cell. This is also probably the case for IpaJ in Golgi fragmentation and STING activation and for OspB in modulating cell-to-cell spread via mTOR and ERK/MAPK signaling. Some factors, such as IcsA involved in adhesion and actin-tail formation, may genuinely have two very different biochemical functions, used at different points in the infectious cycle. OspE1/E2 seem involved in adhesion to host cells and stability of tight junctions. However, others may not. At its position at the T3SS needle tip, IpaB is involved in CD44 interactions (Skoudy et al., 2000), host cell sensing, translocon pore formation (Blocker et al., 1999), and directly or indirectly, and lysis of the single and double membrane entry vacuoles (High et al., 1992; Page et al., 1999). Once secreted, IpaB plays a role in macrophage pyroptosis (Guichon et al., 2001) and cell cycle arrest (Iwai et al., 2007). It is difficult to comprehend how IpaB could exert all these functions without possessing multiple biological activities, which remain unclear. However, some of these functions may have been misattributed to IpaB due to pleotropic effects stemming from its involvement in translocon formation, which is crucial for Shigella entry.
Concluding Remarks
The vast majority of the functional work presented here has been done on S. flexneri. Comparative analysis of the dominant Shigella genomes causing disease is required for further analysis of virulence determinants, such as any involved lysis of the single and double membrane entry vacuoles, which remain completely unclear, and of their epidemiological consequences. The importance of such genomic investigations has recently been highlighted (The et al., 2016). Identification of important and conserved effectors and other virulence determinants that cause disease will contribute to an overall understanding of infection, further illuminating species tropism and transmission and helping to create a pan-Shigella vaccine, which so far has been unsuccessful.
Author Contributions
EM planned, researched, wrote, and subsequently also formatted the manuscript for publication, assisted by discussions, analysis of published works and editing with AB. EM also generated Figures 5–9.
Funding
AB is the recipient of an Investigator Award (WT104634AIA) from the UK Wellcome Trust. EM was an undergraduate student in the School of Cellular and Molecular Medicine at the University of Bristol, UK.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank A. Dorothea Roehrich for providing Figures 1–4, originally published in her PhD thesis, and apologize in advance to any colleague whose work could not be cited because of space constraints.
References
Al-Hasani, K., Henderson, I. R., Sakellaris, H., Rajakumar, K., Grant, T., Nataro, J. P., et al. (2000). The sigA gene which is borne on the she pathogenicity island of Shigella flexneri 2a encodes an exported cytopathic protease involved in intestinal fluid accumulation. Infect. Immun. 68, 2457–2463. doi: 10.1128/IAI.68.5.2457-2463.2000
PubMed Abstract | CrossRef Full Text | Google Scholar
Al-Hasani, K., Navarro-Garcia, F., Huerta, J., Sakellaris, H., and Adler, B. (2009). The immunogenic SigA enterotoxin of Shigella flexneri 2a binds to HEp-2 cells and induces fodrin redistribution in intoxicated epithelial cells. PLoS ONE 4:e8223. doi: 10.1371/journal.pone.0008223
PubMed Abstract | CrossRef Full Text | Google Scholar
Allaoui, A., Mounier, J., Prevost, M.-C., Sansonetti, P. J., and Parsot, C. (1992). IcsB: a Shigella flexneri virulence gene necessary for the lysis of protrusions during intercellular spread. Mol. Microbiol. 6, 1605–1616. doi: 10.1111/j.1365-2958.1992.tb00885.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Allison, G. E., and Verma, N. K. (2000). Serotype-converting bacteriophages and O-antigen modification in Shigella flexneri. Trends Microbiol. 8, 17–23. doi: 10.1016/S0966-842X(99)01646-7
PubMed Abstract | CrossRef Full Text | Google Scholar
Alto, N. M., Shao, F., Lazar, C. S., Brost, R. L., Chua, G., Mattoo, S., et al. (2006). Identification of a bacterial type III effector family with G protein mimicry functions. Cell 124, 133–145. doi: 10.1016/j.cell.2005.10.031
PubMed Abstract | CrossRef Full Text | Google Scholar
Ambrosi, C., Pompili, M., Scribano, D., Limongi, D., Petrucca, A., Cannavacciuolo, S., et al. (2014). The Shigella flexneri OspB effector: an early immunomodulator. Int. J. Med. Microbiol. 305, 75–84. doi: 10.1016/j.ijmm.2014.11.004
PubMed Abstract | CrossRef Full Text | Google Scholar
Arbibe, L., Kim, D. W., Batsche, E., Pedron, T., Mateescu, B., Muchardt, C., et al. (2007). An injected bacterial effector targets chromatin access for transcription factor NF-kB to alter transcription of host genes involved in immune responses. Nat. Immunol. 8, 47–56. doi: 10.1038/ni1423
CrossRef Full Text | Google Scholar
Ashida, H., Kim, M., Schmidt-Supprian, M., Ma, A., Ogawa, M., and Sasakawa, C. (2010). A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKy to dampen the host NF-kB-mediated inflammatory response. Nat. Cell Biol. 12, 66–69. doi: 10.1038/ncb2006
CrossRef Full Text | Google Scholar
Ashida, H., Nakano, H., and Sasakawa, C. (2013). Shigella IpaH0722 E3 ubiquitin ligase effector targets TRAF2 to inhibit PKC–NF-kB activity in invaded epithelial cells. PLoS Pathog. 9:e1003409. doi: 10.1371/journal.ppat.1003409
PubMed Abstract | CrossRef Full Text | Google Scholar
Ashida, H., Ogawa, M., Mimuro, H., Kobayashi, T., Sanada, T., and Sasakawa, C. (2011). Shigella are versatile mucosal pathogens that circumvent the host innate immune system. Curr. Opin. Immunol. 23, 448–455. doi: 10.1016/j.coi.2011.06.001
PubMed Abstract | CrossRef Full Text | Google Scholar
Ashida, H., Toyotome, T., Nagai, T., and Sasakawa, C. (2007). Shigella chromosomal IpaH proteins are secreted via the type III secretion system and act as effectors. Mol. Microbiol. 63, 680–693. doi: 10.1111/j.1365-2958.2006.05547.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Barta, M. L., Guragain, M., Adam, P., Dickenson, N. E., Patil, M., Geisbrecht, B. V., et al. (2012). Identification of the bile salt binding site on IpaD from Shigella flexneri and the influence of ligand binding on IpaD structure. Proteins 80, 935–945. doi: 10.1002/prot.23251
PubMed Abstract | CrossRef Full Text | Google Scholar
Bârzu, S., Benjelloun-Touimi, Z., Phalipon, A., Sansonetti, P., and Parsot, C. (1997). Functional analysis of the Shigella flexneri IpaC invasin by insertional mutagenesis. Infect. Immun. 65, 1599–1605.
PubMed Abstract | Google Scholar
Baxt, L. A., and Goldberg, M. B. (2014). Host and bacterial proteins that repress recruitment of LC3 to Shigella early during infection. PLoS ONE 9:e94653. doi: 10.1371/journal.pone.0094653
PubMed Abstract | CrossRef Full Text | Google Scholar
Benjelloun-Touimi, Z., Sansonetti, P. J., and Parsot, C. (1995). SepA, the major extracellular protein of Shigella flexneri: autonomous secretion and involvement in tissue invasion. Mol. Microbiol. 17, 123–135. doi: 10.1111/j.1365-2958.1995.mmi_17010123.x
PubMed Abstract | CrossRef Full Text
Bergounioux, J., Elisee, R., Prunier, A.-L., Donnadieu, F., Sperandio, B., Sansonetti, P., et al. (2012). Calpain activation by the Shigella flexneri effector VirA regulates key steps in the formation and life of the bacterium's epithelial niche. Cell Host Microbe 11, 240–252. doi: 10.1016/j.chom.2012.01.013
PubMed Abstract | CrossRef Full Text | Google Scholar
Bernardini, M. L., Mounier, J., D'Hauteville, H., Coquis-Rondon, M., and Sansonetti, P. J. (1989). Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. U.S.A. 86, 3867–3871.
PubMed Abstract | Google Scholar
Beuzón, C. R., Meresse, S., Unsworth, K. E., Ruiz-Albert, J., Garvis, S., Waterman, S. R., et al. (2000). Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19, 3235–3249. doi: 10.1093/emboj/19.13.3235
PubMed Abstract | CrossRef Full Text | Google Scholar
Blocker, A., Gounon, P., Larquet, E., Niebuhr, K., Cabiaux, V., Parsot, C., et al. (1999). The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J. Cell Biol. 147, 683–693. doi: 10.1083/jcb.147.3.683
PubMed Abstract | CrossRef Full Text | Google Scholar
Bourdet-Sicard, R., Rudiger, M., Jockusch, B. M., Gounon, P., Sansonetti, P. J., and Nhieu, G. T. (1999). Binding of the Shigella protein IpaA to vinculin induces F-actin depolymerization. EMBO J. 18, 5853–5862. doi: 10.1093/emboj/18.21.5853
PubMed Abstract | CrossRef Full Text | Google Scholar
Brandon, L. D., Goehring, N., Janakiraman, A., Yan, A. W., Wu, T., Beckwith, J., et al. (2003). IcsA, a polarly localized autotransporter with an atypical signal peptide, uses the Sec apparatus for secretion, although the Sec apparatus is circumferentially distributed. Mol. Microbiol. 50, 45–60. doi: 10.1046/j.1365-2958.2003.03674.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Brotcke Zumsteg, A., Goosmann, C., Brinkmann, V., Morona, R., and Zychlinsky, A. (2014). IcsA is a Shigella flexneri adhesin regulated by the type III secretion system and required for pathogenesis. Cell Host Microbe 15, 435–445. doi: 10.1016/j.chom.2014.03.001
PubMed Abstract | CrossRef Full Text | Google Scholar
Buchrieser, C., Glaser, P., Rusniok, C., Nedjari, H., D'Hauteville, H., Kunst, F., et al. (2000). The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 38, 760–771. doi: 10.1046/j.1365-2958.2000.02179.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Burnaevskiy, N., Fox, T. G., Plymire, D., Ertelt, J., Weigele, B. A., Selyunin, A. S., et al. (2013). Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature 496, 106–109. doi: 10.1038/nature12004
PubMed Abstract | CrossRef Full Text | Google Scholar
Burnaevskiy, N., Peng, T., Reddick, L. E., Hang, H. C., and Alto, N. M. (2015). Myristoylome profiling reveals a concerted mechanism of ARF GTPase deacylation by the bacterial protease IpaJ. Mol. Cell. 58, 110–122. doi: 10.1016/j.molcel.2015.01.040
PubMed Abstract | CrossRef Full Text | Google Scholar
Buysse, J. M., Dunyak, D. S., Hartman, A. B., and Venkatesan, M. M. (1997). Identification and molecular characterization of a 27 kDa Shigella flexneri invasion plasmid antigen, IpaJ. Microb. Pathog. 23, 357–369. doi: 10.1006/mpat.1997.0164
PubMed Abstract | CrossRef Full Text | Google Scholar
Campbell-Valois, F.-X., Sachse, M., Sansonetti, P. J., and Parsot, C. (2015). Escape of actively secreting Shigella flexneri from ATG8/LC3-positive vacuoles formed during cell-to-cell spread is facilitated by IcsB and VirA. MBio 6, 1–11. doi: 10.1128/mBio.02567-14
PubMed Abstract | CrossRef Full Text | Google Scholar
Campbell-Valois, F.-X., Schnupf, P., Nigro, G., Sachse, M., Sansonetti, P. J., and Parsot, C. (2014). A fluorescent reporter reveals on/off regulation of the Shigella type III secretion apparatus during entry and cell-to-cell spread. Cell Host Microbe 15, 177–189. doi: 10.1016/j.chom.2014.01.005
PubMed Abstract | CrossRef Full Text | Google Scholar
Canizalez-Roman, A., and Navarro-García, F. (2003). Fodrin CaM-binding domain cleavage by Pet from enteroaggregative Escherichia coli leads to actin cytoskeletal disruption. Mol. Microbiol. 48, 947–958. doi: 10.1046/j.1365-2958.2003.03492.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Carpenter, C. D., Cooley, B. J., Needham, B. D., Fisher, C. R., Trent, M. S., Gordon, V., et al. (2014). The Vps/VacJ ABC transporter is required for intercellular spread of Shigella flexneri. Infect. Immun. 82, 660–669. doi: 10.1128/IAI.01057-13
PubMed Abstract | CrossRef Full Text | Google Scholar
Cohen, D., Green, M. S., Block, C., Slepon, R., and Ofek, I. (1991). Prospective study of the association between serum antibodies to lipopolysaccharide O antigen and the attack rate of Shigellosis. J. Clin. Microbiol. 29, 386–389.
PubMed Abstract | Google Scholar
Coron, E., Flamant, M., Aubert, P., Wedel, T., Pedron, T., Letessier, E., et al. (2009). Characterisation of early mucosal and neuronal lesions following Shigella flexneri infection in human colon. PLoS ONE 4:e4713. doi: 10.1371/journal.pone.0004713
PubMed Abstract | CrossRef Full Text | Google Scholar
De Geyter, C., Vogt, B., Benjelloun-Touimi, Z., Sansonetti, P. J., Ruysschaert, J.-M., Parsot, C., et al. (1997). Purification of IpaC, a protein involved in entry of Shigella flexneri into epithelial cells and characterization of its interaction with lipid membranes. FEBS Lett. 400, 149–154. doi: 10.1016/S0014-5793(96)01379-8
PubMed Abstract | CrossRef Full Text | Google Scholar
Dobbs, N., Burnaevskiy, N., Chen, D., Gonugunta, V. K., Neal, M., Yan, N., et al. (2015). STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18, 157–168. doi: 10.1016/j.chom.2015.07.001
PubMed Abstract | CrossRef Full Text | Google Scholar
Dong, N., Zhu, Y., Lu, Q., Hu, L., Zheng, Y., and Shao, F. (2012). Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell 150, 1029–1041. doi: 10.1016/j.cell.2012.06.050
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, J., Reeves, A. Z., Klein, J. A., Twedt, D. J., Knodler, L. A., and Lesser, C. F. (2016). The type III secretion system apparatus determines the intracellular niche of bacterial pathogens. Proc. Natl. Acad. Sci. U.S.A. 113, 4794–4799. doi: 10.1073/pnas.1520699113
PubMed Abstract | CrossRef Full Text | Google Scholar
Egile, C., Loisel, T. P., Laurent, V., Li, R., Pantaloni, D., Sansonetti, P. J., et al. (1999). Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell Biol. 146, 1319–1332. doi: 10.1083/jcb.146.6.1319
PubMed Abstract | CrossRef Full Text | Google Scholar
Espina, M., Olive, A. J., Kenjale, R., Moore, D. S., Ausar, S. F., Kaminski, R. W., et al. (2006). IpaD localizes to the tip of the type III secretion system needle of Shigella flexneri. Infect. Immun. 74, 4391–4400. doi: 10.1128/IAI.00440-06
PubMed Abstract | CrossRef Full Text | Google Scholar
Faherty, C., Harper, J. M., Shea-Donohue, T., Barry, E. M., Kaper, J. B., Fasano, A., et al. (2012b). Chromosomal and plasmid-encoded factors of Shigella flexneri induce secretogenic activity ex vivo. PLoS ONE 7:e49980. doi: 10.1371/journal.pone.0049980
PubMed Abstract | CrossRef Full Text | Google Scholar
Faherty, C., Redman, J., Rasko, D., Barry, E., and Nataro, J. (2012a). Shigella flexneri effectors OspE1 and OspE3 mediate induced adherence to the colonic epithelium following biles salts exposure. Mol. Microbiol. 85, 107–121. doi: 10.1111/j.1365-2958.2012.08092.x
CrossRef Full Text | Google Scholar
Faherty, C., Wu, T., Morris, C., Grassel, C., Rasko, D., Harper, J., et al. (2016). The synthesis of OspD3 (ShET2) in Shigella flexneri is independent of OspC1. Gut Microbes 7, 486–502. doi: 10.1080/19490976.2016.1239682
PubMed Abstract | CrossRef Full Text | Google Scholar
Farfán, M. J., Toro, C. S., Barry, E. M., and Nataro, J. P. (2011). Shigella enterotoxin-2 is a type III effector that participates in Shigella-induced interleukin 8 secretion by epithelial cells. FEMS Immunol. Med. Microbiol. 61, 332–339. doi: 10.1111/j.1574-695X.2011.00778.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Faruque, S. M., Khan, R., Kamruzzaman, M., Yamasaki, S., Ahmad, Q. S., Azim, T., et al. (2002). Isolation of Shigella dysenteriae type 1 and S. flexneri strains from surface waters in Bangladesh: comparative molecular analysis of environmental Shigella isolates versus clinical Strains. Appl. Environ. Microbiol. 68, 3908–3913. doi: 10.1128/aem.68.8.3908-3913.2002
PubMed Abstract | CrossRef Full Text | Google Scholar
Fasano, A., Noriega, F. R., Liao, F. M., Wang, W., and Levine, M. M. (1997). Effect of shigella enterotoxin 1 (ShET1) on rabbit intestine in vitro and in vivo. Gut 40, 505–511. doi: 10.1136/gut.40.4.505
PubMed Abstract | CrossRef Full Text | Google Scholar
Fasano, A., Noriega, F. R., Maneval, D. R., Chanasongcram, S., Russell, R., Guandalini, S., et al. (1995). Shigella enterotoxin 1: an enterotoxin of Shigella flexneri 2a active in rabbit small intestine in vivo and in vitro. J. Clin. Invest. 95, 2853–2861. doi: 10.1172/JCI117991
PubMed Abstract | CrossRef Full Text | Google Scholar
Fernandez-Prada, C. M., Hoover, D. L., Tall, B. D., Hartman, A. B., Kopelowitz, J., and Venkatesan, M. M. (2000). Shigella flexneri IpaH7.8 facilitates escape of virulent bacteria from the endocytic vacuoles of mouse and human macrophages. Infect. Immun. 68, 3608–3619. doi: 10.1128/IAI.68.6.3608-3619.2000
CrossRef Full Text | Google Scholar
Fontaine, A., Arondel, J., and Sansonetti, P. J. (1988). Role of shiga toxin in the pathogenesis of bacillary dysentery, studied by using a Tox-mutant of Shigella dysenteriae 1. Infect. Immun. 56, 3099–3109.
PubMed Abstract | Google Scholar
Fukuda, I., Suzuki, T., Munakata, H., Hayashi, N., Katayama, E., Yoshikawa, M., et al. (1995). Cleavage of Shigella surface protein VirG occurs at a specific site, but the secretion is not essential for intracellular spreading. J. Bacteriol. 177, 1719–1726. doi: 10.1128/jb.177.7.1719-1726.1995
CrossRef Full Text | Google Scholar
Garza-Mayers, A. C., Miller, K. A., Russo, B. C., Nagda, D. V., and Goldberg, M. B. (2015). Shigella flexneri regulation of ARF6 activation during bacterial entry via an IpgD-mediated positive feedback loop. MBio 6, 1–12. doi: 10.1128/mBio.02584-14
PubMed Abstract | CrossRef Full Text | Google Scholar
Germane, K., Ohi, R., Goldberg, M. B., and Spiller, B. W. (2008). Structural and functional studies indicate that Shigella VirA is not a protease and does not directly destabilize microtubules. Biochemistry 47, 10241–10243. doi: 10.1021/bi801533k
CrossRef Full Text | Google Scholar
Grishin, A. M., Condos, T. E. C., Barber, K. R., Campbell-Valois, F.-X., Parsot, C., Shaw, G. S., et al. (2014). Structural basis for the inhibition of host protein ubiquitination by Shigella effector kinase OspG. Cell Struct. 22, 878–888. doi: 10.1016/j.str.2014.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
Guan, S., Bastin, D. A., and Verma, N. K. (1999). Functional analysis of the O antigen glucosylation gene cluster of Shigella flexneri bacteriophage SfX. Microbiology 145, 1263–1273. doi: 10.1099/13500872-145-5-1263
PubMed Abstract | CrossRef Full Text | Google Scholar
Guichon, A., Hersh, D., Smith, M. R., and Zychlinsky, A. (2001). Structure-function analysis of the Shigella virulence factor IpaB. J. Bacteriol. 183, 1269–1276. doi: 10.1128/JB.183.4.1269-1276.2001
PubMed Abstract | CrossRef Full Text | Google Scholar
Gutierrez-Jimenez, J., Arciniega, I., and Navarro-Garcia, F. (2008). The serine protease motif of Pic mediates a dose-dependent mucolytic activity after binding to sugar constituents of the mucin substrate. Microb. Pathog. 45, 115–123. doi: 10.1016/j.micpath.2008.04.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Hachani, A., Biskri, L., Rossi, G., Marty, A., Menard, R., Sansonetti, P., et al. (2007). IpgB1 and IpgB2, two homologous effectors secreted via the Mxi-Spa type III secretion apparatus, cooperate to mediate polarized cell invasion and inflammatory potential of Shigella flexenri. Microbes Infect. 10, 260–268. doi: 10.1016/j.micinf.2007.11.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Handa, Y., Suzuki, M., Ohya, K., Iwai, H., Ishijima, N., Koleske, A. J., et al. (2007). Shigella IpgB1 promotes bacterial entry through the ELMO-Dock180 machinery. Nat. Cell Biol. 9, 121–128. doi: 10.1038/ncb1526
PubMed Abstract | CrossRef Full Text | Google Scholar
Harrington, S. M., Sheikh, J., Henderson, I. R., Ruiz-perez, F., Cohen, P. S., and Nataro, J. P. (2009). The Pic protease of enteroaggregative Escherichia coli promotes intestinal colonization and growth in the presence of mucin. Infect. Immun. 77, 2465–2473. doi: 10.1128/IAI.01494-08
PubMed Abstract | CrossRef Full Text | Google Scholar
Henderson, I. R., Czeczulin, J., Eslava, C., Noriega, F., and Nataro, J. P. (1999). Characterization of Pic, a secreted protease of Shigella flexneri and enteroaggregative Escherichia coli. Infect. Immun. 67, 5587–5596.
PubMed Abstract | Google Scholar
High, N., Mounier, J., Prevost, M. C., and Sansonetti, P. J. (1992). IpaB of Shigella flexneri causes entry into epithelial cells and escape from the phagocytic vacuole. EMBO J. 11, 1991–1999.
PubMed Abstract | Google Scholar
Hong, M., Gleason, Y., Wyckoff, E. E., and Payne, S. M. (1998). Identification of two Shigella flexneri chromosomal loci involved in intercellular spreading. Infect. Immun. 66, 4700–4710.
PubMed Abstract | Google Scholar
Huang, Z., Sutton, S. E., Wallenfang, A. J., Orchard, R. C., Wu, X., Feng, Y., et al. (2009). Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics. Nat. Struct. Mol. Biol. 16, 853–860. doi: 10.1038/nsmb.1647
PubMed Abstract | CrossRef Full Text | Google Scholar
Ingersoll, M. A., Moss, J. E., Weinrauch, Y., Fisher, P. E., Groisman, E. A., and Zychlinsky, A. (2003). The ShiA protein encoded by the Shigella flexneri SHI-2 pathogenicity island attenuates inflammation. Cell. Microbiol. 5, 797–807. doi: 10.1046/j.1462-5822.2003.00320.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Iwai, H., Kim, M., Yoshikawa, Y., Ashida, H., Ogawa, M., Fujita, Y., et al. (2007). A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell 130, 611–623. doi: 10.1016/j.cell.2007.06.043
PubMed Abstract | CrossRef Full Text | Google Scholar
Izard, T., Tran Van Nhieu, G., and Bois, P. R. J. (2006). Shigella applies molecular mimicry to subvert vinculin and invade host cells. J. Cell Biol. 175, 465–475. doi: 10.1083/jcb.200605091
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalluir, P., Cummings, K. C., Abbott, S., Malcolm, G. B., Hutcheson, K., Beall, A., et al. (2004). Epidemiological features of a newly described serotype of Shigella boydii. Epidemiol. Infect. 132, 579–583. doi: 10.1017/S0950268804002377
CrossRef Full Text | Google Scholar
Kim, D. W., Chu, H., Joo, D. H., Jang, M. S., Choi, J. H., Park, S.-M., et al. (2008). OspF directly attenuates the activity of extracellular signal-regulated kinase during invasion by Shigella flexneri in human dendritic cells. Mol. Immunol. 45, 3295–3301. doi: 10.1016/j.molimm.2008.02.013
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, D. W., Lenzen, G., Page, A.-L., Legrain, P., Sansonetti, P. J., and Parsot, C. (2005). The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. U.S.A. 102, 14046–14051. doi: 10.1073/pnas.0504466102
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, M., Ogawa, M., Fujita, Y., Yoshikawa, Y., Nagai, T., Koyama, T., et al. (2009). Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nat. Lett. 459, 578–582. doi: 10.1038/nature07952
PubMed Abstract | CrossRef Full Text | Google Scholar
Klink, B. U., Barden, S., Heidler, T. V., Borchers, C., Ladwein, M., Stradal, T. E. B., et al. (2010). Structure of Shigella IpgB2 in complex with human RhoA. J. Biol. Chem. 285, 17197–17208. doi: 10.1074/jbc.M110.107953
PubMed Abstract | CrossRef Full Text | Google Scholar
Knodler, L. A., Finlay, B., and Steele-Mortimer, O. (2005). The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J. Biol. Chem. 280, 9058–9064. doi: 10.1074/jbc.M412588200
PubMed Abstract | CrossRef Full Text | Google Scholar
Kobayashi, T., Ogawa, M., Sanada, T., Mimuro, H., Kim, M., Ashida, H., et al. (2013). The Shigella OspC3 effector inhibits Caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 13, 570–583. doi: 10.1016/j.chom.2013.04.012
PubMed Abstract | CrossRef Full Text | Google Scholar
Konradt, C., Frigimelica, E., Nothelfer, K., Puhar, A., Salgado-Pabon, W., Bartolom, V. D., et al. (2011). The Shigella flexneri type three secretion system effector IpgD tnhibits T Cell migration by manipulating host phosphoinositide metabolism. Cell Host Microbe 9, 263–272. doi: 10.1016/j.chom.2011.03.010
PubMed Abstract | CrossRef Full Text | Google Scholar
Kotloff, K. L., Pasetti, M. F., Barry, E. M., Nataro, J. P., Wasserman, S. S., Sztein, M. B., et al. (2004). Deletion in the Shigella enterotoxin genes further attenuates Shigella flexneri 2a bearing guanine auxotrophy in a phase 1 trial of CVD 1204 and CVD 1208. J. Infect. Dis. 190, 1745–1754. doi: 10.1086/424680
PubMed Abstract | CrossRef Full Text | Google Scholar
Kuwae, A., Yoshida, S., Tamano, K., Mimuro, H., Suzuki, T., and Sasakawa, C. (2001). Shigella invasion of macrophage requires the insertion of IpaC into the host plasma membrane. J. Biol. Chem. 276, 32230–32239. doi: 10.1074/jbc.M103831200
PubMed Abstract | CrossRef Full Text | Google Scholar
Lafont, F., Tran Van Nhieu, G., Hanada, K., Sansonetti, P., and van der Goot, F. G. (2002). Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44-IpaB interaction. EMBO J. 21, 4449–4457. doi: 10.1093/emboj/cdf457
PubMed Abstract | CrossRef Full Text | Google Scholar
Levine, M. M., Kotloff, K. L., Barry, E. M., Pasetti, M. F., and Sztein, M. B. (2013). Clinical trials of Shigella vaccines: two steps forward and one step back on a long, hard road. Nat. Rev. Microbiol. 5, 540–553. doi: 10.1038/nrmicro1662
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, H., Xu, H., Zhou, Y., Zhang, J., Long, C., Li, S., et al. (2007). The phosphothreonine lyase activity of a bacterial type III effector family. Science 315, 1000–1003. doi: 10.1126/science.1138960
PubMed Abstract | CrossRef Full Text | Google Scholar
Lu, R., Herrera, B. B., Eshleman, H. D., Fu, Y., Bloom, A., Li, Z., et al. (2015). Shigella effector OspB activates mTORC1 in a manner that depends on IQGAP1 and promotes cell proliferation. PLoS Pathog. 11:e1005200. doi: 10.1371/journal.ppat.1005200
PubMed Abstract | CrossRef Full Text | Google Scholar
Maejima, I., Takahashi, A., Omori, H., Kimura, T., Takabatake, Y., Saitoh, T., et al. (2013). Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 32, 2336–2347. doi: 10.1038/emboj.2013.171
PubMed Abstract | CrossRef Full Text | Google Scholar
Martinez-Argudo, I., and Blocker, A. J. (2010). The Shigella T3SS needle transmits a signal for MxiC release, which controls secretion of effectors. Mol. Microbiol. 78, 1365–1378. doi: 10.1111/j.1365-2958.2010.07413.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Mayer, C. L., Leibowitz, C. S., Kurosawa, S., and Stearns-kurosawa, D. J. (2012). Shiga toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals. Toxins 4, 1261–1287. doi: 10.3390/toxins4111261
PubMed Abstract | CrossRef Full Text | Google Scholar
Mayo, L. D., and Donner, D. B. (2001). A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. U.S.A. 98, 11598–11603. doi: 10.1073/pnas.181181198
PubMed Abstract | CrossRef Full Text | Google Scholar
Mazurkiewicz, P., Thomas, J., Thompson, J. A., Liu, M., Arbibe, L., Sansonetti, P., et al. (2008). SpvC is a Salmonella effector with phosphothreonine lyase activity on host mitogen-activated protein kinases. Mol. Microbiol. 67, 1371–1383. doi: 10.1111/j.1365-2958.2008.06134.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Mellouk, N., Weiner, A., Aulner, N., Schmitt, C., Elbaum, M., Shorte, S. L., et al. (2014). Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe 16, 517–530. doi: 10.1016/j.chom.2014.09.005
PubMed Abstract | CrossRef Full Text | Google Scholar
Menard, R., Prevostt, M., Gounont, P., Sansonetti, P., and Dehio, C. (1996). The secreted Ipa complex of Shigella flexneri promotes entry into mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 93, 1254–1258. doi: 10.1073/pnas.93.3.1254
PubMed Abstract | CrossRef Full Text | Google Scholar
Miura, M., Terajima, J., Izumiya, H., Mitobe, J., Komano, T., and Watanabe, H. (2006). OspE2 of Shigella sonnei is required for the maintenance of cell architecture of bacterium-infected cells. Infect. Immun. 74, 2587–2595. doi: 10.1128/IAI.74.5.2587-2595.2006
PubMed Abstract | CrossRef Full Text | Google Scholar
Monack, D. M., and Theriot, J. A. (2001). Actin-based motility is sufficient for bacterial membrane protrusion formation and host cell uptake. Cell. Microbiol. 3, 633–647. doi: 10.1046/j.1462-5822.2001.00143.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Mounier, J., Boncompain, G., Senerovic, L., Lagache, T., Chrétien, F., Perez, F., et al. (2012). Shigella effector IpaB-induced cholesterol relocation disrupts the Golgi complex and recycling network to inhibit host cell secretion. Cell Host Microbe 12, 381–389. doi: 10.1016/j.chom.2012.07.010
PubMed Abstract | CrossRef Full Text | Google Scholar
Mounier, J., Popoff, M. R., Enninga, J., Frame, M. C., Sansonetti, P., and Tran Van Nhieu, G. (2009). The IpaC carboxyterminal effector domain mediates Src-dependent actin polymerization during Shigella invasion of epithelial cells. PLoS Pathog. 5:e1000271. doi: 10.1371/journal.ppat.1000271
PubMed Abstract | CrossRef Full Text | Google Scholar
Murillo, I., Martinez-Argudo, I., and Blocker, A. J. (2016). Genetic dissection of the signaling cascade that controls activation of the Shigella type III secretion system from the needle tip. Sci. Rep. 6:27649. doi: 10.1038/srep27649
PubMed Abstract | CrossRef Full Text | Google Scholar
Murphy, K. (2012). Janeway's Immunobiology, 8th Edn. New York, NY: Garland Science.
Google Scholar
Nadler, C., Baruch, K., Kobi, S., Mills, E., Haviv, G., Farago, M., et al. (2010). The type III secretion effector NleE inhibits NF-kB activation. PLoS Pathog. 6:e1000743. doi: 10.1371/journal.ppat.1000743
CrossRef Full Text | Google Scholar
Nataro, J. P., Seriwatana, J., Fasano, A., Maneval, D. R., Guers, L. D., Noriega, F., et al. (1995). Identification and cloning of a novel plasmid-encoded enterotoxin of enteroinvasive Escherichia coli and Shigella strains. Infect. Immun. 63, 4721–4728.
PubMed Abstract | Google Scholar
Navarro-Garcia, F., Gutierrez-Jimenez, J., Garcia-Tovar, C., Castro, L. A., Salazar-Gonzalez, H., and Cordova, V. (2010). Pic, an autotransporter protein secreted by different pathogens in the Enterobacteriaceae family, is a potent mucus secretagogue. Infect. Immun. 78, 4101–4109. doi: 10.1128/IAI.00523-10
PubMed Abstract | CrossRef Full Text | Google Scholar
Newton, H. J., Pearson, J. S., Badea, L., Kelly, M., Lucas, M., Holloway, G., et al. (2010). The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kB p65. PLoS Pathog. 6:e1000898. doi: 10.1371/journal.ppat.1000898
CrossRef Full Text | Google Scholar
Niebuhr, K., Giuriato, S., Pedron, T., Philpott, D. J., Gaits, F., Sable, J., et al. (2002). Conversion of PtdIns(4,5)P2 into PtdIns(5)P by the S. flexneri effector IpgD reorganizes host cell morphology. EMBO J. 21, 5069–5078. doi: 10.1093/emboj/cdf522
CrossRef Full Text | Google Scholar
Niebuhr, K., Jouihri, N., Allaoui, A., Gounon, P., Sansonetti, P. J., and Parsot, C. (2000). IpgD, a protein secreted by the type III secretion machinery of Shigella flexneri, is chaperoned by IpgE and implicated in entry focus formation. Mol. Microbiol. 38, 8–19. doi: 10.1046/j.1365-2958.2000.02041.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Nishide, A., Kim, M., Takagi, K., Himeno, A., Sanada, T., Sasakawa, C., et al. (2013). Structural basis for the recognition of Ubc13 by the Shigella flexneri effector OspI. J. Mol. Biol. 425, 2623–2631. doi: 10.1016/j.jmb.2013.02.037
PubMed Abstract | CrossRef Full Text | Google Scholar
Nothelfer, K., Arena, E. T., Pinaud, L., Neunlist, M., Mozeleski, B., Belotserkovsky, I., et al. (2014). B lymphocytes undergo TLR2-dependent apoptosis upon Shigella infection. J. Exp. Med. 211, 1215–1229. doi: 10.1084/jem.20130914
PubMed Abstract | CrossRef Full Text | Google Scholar
Ogawa, M., Suzuki, T., Tatsuno, I., Abe, H., and Sasakawa, C. (2003). IcsB, secreted via the type III secretion system, is chaperoned by IpgA and required at the post-invasion stage of Shigella pathogenicity. Mol. Microbiol. 48, 913–931. doi: 10.1046/j.1365-2958.2003.03489.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Ogawa, M., Yoshimori, T., Suzuki, T., Sagara, H., Mizushima, N., and Sasakawa, C. (2005). Escape of intracellular Shigella from autophagy. Science 307, 727–730. doi: 10.1126/science.1106036
PubMed Abstract | CrossRef Full Text | Google Scholar
Ohya, K., Handa, Y., Ogawa, M., Suzuki, M., and Sasakawa, C. (2005). IpgB1 is a novel Shigella effector protein involved in bacterial invasion of host cells. J. Biol. Chem. 280, 24022–24034. doi: 10.1074/jbc.M502509200
PubMed Abstract | CrossRef Full Text | Google Scholar
Okuda, J., Toyotome, T., Kataoka, N., Ohno, M., Abe, H., Shimura, Y., et al. (2005). Shigella effector IpaH9.8 binds to a splicing factor U2AF35 to modulate host immune responses. Biochem. Biophys. Res. Commun. 333, 531–539. doi: 10.1016/j.bbrc.2005.05.145
CrossRef Full Text | Google Scholar
Osiecki, J. C., Barker, J., Picking, W. L., Barnoski Serfis, A., Berring, E., Shah, S., et al. (2001). IpaC from Shigella and SipC from Salmonella possess similar biochemical properties but are functionally distinct. Mol. Microbiol. 42, 469–481. doi: 10.1046/j.1365-2958.2001.02654.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Paetzold, S., Lourido, S., Raupach, B., and Zychlinsky, A. (2007). Shigella flexneri phagosomal escape is independent of invasion. Infect. Immun. 75, 4826–4830. doi: 10.1128/IAI.00454-07
PubMed Abstract | CrossRef Full Text | Google Scholar
Page, A.-L., Ohayon, H., Sansonetti, P. J., and Parsot, C. (1999). The secreted IpaB and IpaC invasins and their cytoplasmic chaperone IpgC are required for intercellular dissemination of Shigella flexneri. Cell. Microbiol. 1, 183–193. doi: 10.1046/j.1462-5822.1999.00019.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Parsot, C., Ageron, E., Penno, C., Mavris, M., Jamoussi, K., Hauteville, H., et al. (2005). A secreted anti-activator, OspD1, and its chaperone, Spa15, are involved in the control of transcription by the type III secretion apparatus activity in Shigella flexneri. Mol. Microbiol. 56, 1627–1635. doi: 10.1111/j.1365-2958.2005.04645.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Pendaries, C., Arbibe, L., Mounier, J., Gozani, O., Cantley, L., Fry, M. J., et al. (2006). PtdIns(5)P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 25, 1024–1034. doi: 10.1038/sj.emboj.7601001
CrossRef Full Text | Google Scholar
Perrett, C. A., Lin, D. Y.-W., and Zhou, D. (2011). Interactions of bacterial proteins with host eukaryotic ubiquitin pathways. Front. Microbiol. 2:143. doi: 10.3389/fmicb.2011.00143
PubMed Abstract | CrossRef Full Text | Google Scholar
Pruneda, J. N., Donelson Smith, F., Daurie, A., Swaney, D. L., Villén, J., Scott, J. D., et al. (2014). E2~Ub conjugates regulate the kinase activity of Shigella effector OspG during pathogenesis. EMBO J. 33, 437–449. doi: 10.1002/embj.201386386
PubMed Abstract | CrossRef Full Text | Google Scholar
Purdy, G. E., and Payne, S. M. (2001). The SHI-3 iron transport island of Shigella boydii 0-1392 carries the genes for aerobactin synthesis and transport. J. Bacteriol. 183, 4176–4182. doi: 10.1128/JB.183.14.4176-4182.2001
PubMed Abstract | CrossRef Full Text | Google Scholar
Ramarao, N., Le Clainche, C., Izard, T., Bourdet-Sicard, R., Ageron, E., Sansonetti, P. J., et al. (2007). Capping of actin filaments by vinculin activated by the Shigella IpaA carboxyl-terminal domain. FEBS Lett. 581, 853–857. doi: 10.1016/j.febslet.2007.01.057
PubMed Abstract | CrossRef Full Text | Google Scholar
Reiterer, V., Grossniklaus, L., Tschon, T., Kasper, C. A., Sorg, I., and Arrieumerlou, C. (2011). Shigella flexneri type III secreted effector OspF reveals new crosstalks of pro-inflammatory signaling pathways during bacterial infection. Cell. Signal. 23, 1188–1196. doi: 10.1016/j.cellsig.2011.03.006
CrossRef Full Text | Google Scholar
Roehrich-Doenitz, A. D., Guillossou, E., Blocker, A. J., and Martinez-Argudo, I. (2013). Shigella IpaD has a dual role: signal transduction from the type III secretion system needle tip and intracellular secretion regulation. Mol. Microbiol. 87, 690–706. doi: 10.1111/mmi.12124
CrossRef Full Text | Google Scholar
Roerich-Doenitz, A. D. (2013). Regulation of Type III Secretion Hierachy in Shigella flexneri. Ph.D. thesis. University of Bristol.
Rohde, J. R., Breitkreutz, A., Chenal, A., Sansonetti, P. J., and Parsot, C. (2007). Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe 1, 77–83. doi: 10.1016/j.chom.2007.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
Rohmer, L., Jacobs, M. A., Brittnacher, M. J., Fong, C., Hayden, H. S., Hocquet, D., et al. (2014). Genomic analysis of the emergence of 20th century epidemic dysentery. BMC Genomics. 15:355. doi: 10.1186/1471-2164-15-355
PubMed Abstract | CrossRef Full Text | Google Scholar
Russo, B. C., Stamm, L. M., Raaben, M., Kim, C. M., Robinson, L. R., Bose, S., et al. (2016). Intermediate filaments enable pathogen docking to trigger type 3 effector translocation. Nat. Microbiol. 7, 1–24. doi: 10.1038/nmicrobiol.2016.25
CrossRef Full Text | Google Scholar
Sack, D. A., Shamsul Hoque, A. T. M., Huq, A., and Etheridge, M. (1994). Is protection against shigellosis induced by natural infection with Plesiomonas shigelloides? Lancet. 343, 1413–1415. doi: 10.1016/S0140-6736(94)92531-3
PubMed Abstract | CrossRef Full Text | Google Scholar
Salgado-Pabón, W., Celli, S., Arena, E. T., Nothelfer, K., Roux, P., Sellge, G., et al. (2013). Shigella impairs T lymphocyte dynamics in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, 4458–4463. doi: 10.1073/pnas.1300981110
PubMed Abstract | CrossRef Full Text | Google Scholar
Sanada, T., Kim, M., Mimuro, H., Suzuki, M., Ogawa, M., Oyama, A., et al. (2012). The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature 483, 623–626. doi: 10.1038/nature10894
PubMed Abstract | CrossRef Full Text | Google Scholar
Sandlin, R. C., Lampel, K. A., Keasler, S. P., Goldberg, M. B., Stolzer, A. L., and Maurelli, A. T. (1995). Avirulence of rough mutants of Shigella flexneri: requirement of O antigen for correct unipolar localization of IcsA in the bacterial outer membrane. Infect. Immun. 63, 229–237.
PubMed Abstract | Google Scholar
Sansonetti, P. J., Ryter, A., Clerc, P., Maurelli, A. T., and Mounier, J. (1986). Multiplication of Shigella flexneri within HeLa Cells: lysis of the phagocytic vacuole and plasmid-mediated contact hemolysis. Infect. Immun. 51, 461–469.
PubMed Abstract | Google Scholar
Schroeder, G. N., and Hilbi, H. (2008). Molecular pathogenesis of Shigella spp.: controlling host cell signalling, invasion, and death by type III secretion. Clin. Microbiol. Rev. 21, 134–156. doi: 10.1128/cmr.00032-07
CrossRef Full Text | Google Scholar
Senerovic, L., Tsunoda, S. P., Goosmann, C., Brinkmann, V., Zychlinsky, A., Meissner, F., et al. (2012). Spontaneous formation of IpaB ion channels in host cell membranes reveals how Shigella induces pyroptosis in macrophages. Cell Death Dis. 3, 1–10. doi: 10.1038/cddis.2012.124
PubMed Abstract | CrossRef Full Text | Google Scholar
Seyedarabi, A., Sullivan, J. A., Sasakawa, C., and Pickersgill, R. W. (2010). A disulfide driven domain swap switches off the activity of Shigella IpaH9.8 E3 ligase. FEBS Lett. 584, 4163–4168. doi: 10.1016/j.febslet.2010.09.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Shands, J. W., and Chun, P. W. (1980). The dispersion of Gram-negative lipopolysaccharide by deoxycholate. J. Biol. Chem. 255, 1221–1226.
PubMed Abstract | Google Scholar
Shepherd, J. G., Wang, L., and Reeves, P. R. (2000). Comparison of O-antigen gene clusters of Escherichia coli (Shigella) sonnei and Plesiomonas shigelloides O17: Sonnei gained its current plasmid-borne O-antigen genes from P. shigelloides in a recent event. Infect. Immun. 68, 6056–6061. doi: 10.1128/iai.68.10.6056-6061.2000
PubMed Abstract | CrossRef Full Text | Google Scholar
Shibata, T., Takeshima, F., Chen, F., Alt, F. W., and Snapper, S. B. (2002). Cdc42 facilitates invasion but not the actin-based motility of Shigella. Curr. Biol. 12, 341–345. doi: 10.1016/S0960-9822(02)00689-9
CrossRef Full Text | Google Scholar
Skoudy, A., Mounier, J., Aruffo, A., Ohayon, H., Gounon, P., Sansonetti, P., et al. (2000). CD44 binds to the Shigella IpaB protein and participates in bacterial invasion of epithelial cells. Cell. Microbiol. 2, 19–33. doi: 10.1046/j.1462-5822.2000.00028.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Sperandio, B., Regnault, B., Guo, J., Zhang, Z., Stanley, Jr., S. L., Sansonetti, P. J., et al. (2008). Virulent Shigella flexneri subverts the host innate immune response through manipulation of antimicrobial peptide gene expression. J. Exp. Med. 205, 1121–1132. doi: 10.1084/jem.20071698
PubMed Abstract | CrossRef Full Text | Google Scholar
Steele-Mortimer, O., Knodler, L. A., Marcus, S. L., Scheid, M. P., Goh, B., Pfeifer, C. G., et al. (2000). Activation of Akt/protein kinase B in epithelial cells by the Salmonella typhimurium effector SigD. J. Biol. Chem. 275, 37718–37724. doi: 10.1074/jbc.M008187200
PubMed Abstract | CrossRef Full Text | Google Scholar
Stuart, L. M., Paquette, N., and Boyer, L. (2013). Effector-triggered versus pattern-triggered immunity: how animals sense pathogens. Nat. Rev. Immunol. 13, 199–206. doi: 10.1038/nri3398
PubMed Abstract | CrossRef Full Text | Google Scholar
Suzuki, S., Franchi, L., He, Y., Munoz-Planillo, R., Mimuro, H., Suzuki, T., et al. (2014b). Shigella type III secretion protein MxiI is recognized by Naip2 to induce Nlrc4 inflammasome activation independently of Pkcδ. PLoS Pathog. 10:e1003926. doi: 10.1371/journal.ppat.1003926
PubMed Abstract | CrossRef Full Text | Google Scholar
Suzuki, S., Mimuro, H., Kim, M., Ogawa, M., Ashida, H., Toyotome, T., et al. (2014a). Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc. Natl. Acad. Sci. U.S.A. 4254–4263. doi: 10.1073/pnas.1324021111
PubMed Abstract | CrossRef Full Text | Google Scholar
Suzuki, T., Murai, T., Tobe, T., Yoshikawa, M., and Sasakawa, C. (1994). Identification and characterization of a chromosomal virulence gene, vacJ, required for intercellular spreading of Shigella flexneri. Mol. Microbiol. 11, 31–41. doi: 10.1111/j.1365-2958.1994.tb00287.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Terry, C. M., Picking, W. L., Birket, S. E., Flentie, K., Hoffman, B. M., Barker, J. R., et al. (2008). The C-terminus of IpaC is required for effector activities related to Shigella invasion of host cells. Microb. Pathog. 45, 282–289. doi: 10.1016/j.micpath.2008.06.003
PubMed Abstract | CrossRef Full Text | Google Scholar
The, H. C., Thanh, D. P., Holt, K. E., Thomson, N. R., and Baker, S. (2016). The genomic signatures of Shigella evolution, adaptation and geographical spread. Nat. Rev. Microbiol. 14, 235–250. doi: 10.1038/nrmicro.2016.10
PubMed Abstract | CrossRef Full Text | Google Scholar
Thirumalai, K., Kim, K., and Zychlinsky, A. (1997). IpaB, a Shigella flexneri invasin, colocalizes with interleukin-1β-converting enzyme in the cytoplasm of macrophages. Infect. Immun. 65, 787–793.
Google Scholar
Tran Van Nhieu, G., Ben-Ze'ev, A., and Sansonetti, P. J. (1997). Modulation of bacterial entry into epithelial cells by association between vinculin and the Shigella IpaA invasin. EMBO J. 16, 2717–2729.
PubMed Abstract | Google Scholar
Tran Van Nhieu, G., Caron, E., Hall, A., and Sansonetti, P. J. (1999). IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J. 18, 3249–3262. doi: 10.1093/emboj/18.12.3249
PubMed Abstract | CrossRef Full Text | Google Scholar
Uchiya, K., Tobe, T., Komatsu, K., Suzuki, T., Watarai, M., Fukuda, I., et al. (1995). Identification of a novel virulence gene, virA, on the large plasmid of Shigella, involved in invasion and intercellular spreading. Mol. Microbiol. 17, 241–250. doi: 10.1111/j.1365-2958.1995.mmi_17020241.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Vargas, M., Gascon, J., Jimenez De Anta, M. T., and Vila, J. (1999). Prevalence of Shigella enterotoxins 1 and 2 among Shigella strains isolated from patients with traveller's diarrhea. J. Clin. Microbiol. 37, 3608–3611.
Google Scholar
Veenendaal, A. K. J., Hodgkinson, J. L., Schwarzer, L., Stabat, D., Zenk, S. F., and Blocker, A. J. (2007). The type III secretion system needle tip complex mediates host cell sensing and translocon insertion. Mol. Microbiol. 63, 1719–1730. doi: 10.1111/j.1365-2958.2007.05620.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Vokes, S. A., Reeves, S. A., Torres, A. G., and Payne, S. M. (1999). The aerobactin iron transport system genes in Shigella flexneri are present within a pathogenicity island. Mol. Microbiol. 33, 63–73. doi: 10.1046/j.1365-2958.1999.01448.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiner, A., Mellouk, N., Lopez-Montero, N., Chang, Y.-Y., Souque, C., Schmitt, C., et al. (2016). Macropinosomes are key players in early Shigella invasion and vacuolar escape in epithelial cells. PLoS Pathog. 5:e1005602. doi: 10.1371/journal.ppat.1005602
CrossRef Full Text | Google Scholar
West, N. P., Sansonetti, P., Mounier, J., Exley, R. M., Parsot, C., Guadagnini, S., et al. (2005). Optimization of virulence functions through glucosylation of Shigella LPS. Science 307, 1313–1318. doi: 10.1126/science.1108472
PubMed Abstract | CrossRef Full Text | Google Scholar
WHO (2005). Guidelines for the Control of Shigellosis, Including Epidemics due to Shigella dysenteriae Type 1. Geneva.
Woodward, D. L., Clark, C. G., Caldeira, R. A., Ahmed, R., Soule, G., Bryden, L., et al. (2005). Identification and characterization of Shigella boydii 20 serovar nov., a new and emerging Shigella serotype. J. Med. Microbiol. 54, 741–748. doi: 10.1099/jmm.0.46095-0
PubMed Abstract | CrossRef Full Text | Google Scholar
Yang, F., Yang, J., Zhang, X., Chen, L., Jiang, Y., Yan, Y., et al. (2005). Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res. 33, 6445–6458. doi: 10.1093/nar/gki954
PubMed Abstract | CrossRef Full Text | Google Scholar
Yang, J., Zhao, Y., Shi, J., and Shao, F. (2013). Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc. Natl. Acad. Sci.U.S.A. 110, 14408–14413. doi: 10.1073/pnas.1306376110
PubMed Abstract | CrossRef Full Text | Google Scholar
Yavzori, M., Cohen, D., and Orr, N. (2002). Prevalence of the genes for shigella enterotoxins 1 and 2 among clinical isolates of shigella in Israel. Epidemiol. Infect. 128, 533–535. doi: 10.1017/S0950268802006866
PubMed Abstract | CrossRef Full Text | Google Scholar
Yoshida, S., Katayama, E., Kuwae, A., Mimuro, H., Suzuki, T., and Sasakawa, C. (2002). Shigella deliver an effector protein to trigger host microtubule destabilization, which promotes Rac1 activity and efficient bacterial internalization. EMBO J. 21, 2923–2935. doi: 10.1093/emboj/cdf319
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhou, Y., Dong, N., Hu, L., and Shao, F. (2013). The Shigella type three secretion system effector OspG directly and specifically binds to host ubiquitin for activation. PLoS ONE 8:e57558. doi: 10.1371/journal.pone.0057558
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhu, Y., Li, H., Long, C., Hu, L., Xu, H., Liu, L., et al. (2007). Structural insights into the enzymatic mechanism of the pathogenic MAPK phosphothreonine lyase. Mol. Cell. 28, 899–913. doi: 10.1016/j.molcel.2007.11.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Zoppino, F. C. M., Militello, R. D., Slavin, I., Álvarez, C., and Colombo, M. I. (2010). Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic 11, 1246–1261. doi: 10.1111/j.1600-0854.2010.01086.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Zurawski, D. V., Mitsuhata, C., Mumy, K. L., McCormick, B. A., and Maurelli, A. T. (2006). OspF and OspC1 Are Shigella flexneri type III secretion system effectors that are required for post-invasion aspects of virulence. Infect. Immun. 74, 5964–5976. doi: 10.1128/IAI.00594-06
CrossRef Full Text | Google Scholar
Zurawski, D. V., Mumy, K. L., Badea, L., Prentice, J. A., Hartland, E. L., McCormick, B. A., et al. (2008). The NleE/OspZ family of effector proteins is required for polymorphonuclear transepithelial migration, a characteristic shared by enteropathogenic Escherichia coli and Shigella flexneri infections. Infect. Immun. 76, 369–379. doi: 10.1128/IAI.00684-07
PubMed Abstract | CrossRef Full Text | Google Scholar
Zurawski, D. V., Mumy, K. L., Faherty, C. S., McCormick, B. A., and Maurelli, A. T. (2009). Shigella flexneri T3SS effectors OspB and OspF target the nucleus to down-regulate the host inflammatory response via interactions with retinoblastoma protein. Mol. Microbiol. 71, 350–368. doi: 10.1111/j.1365-2958.2008.06524.x
CrossRef Full Text | Google Scholar
Label The Drawing Of An Enteric Bacterium With The Antigens And Virulence Factors.
Source: https://www.frontiersin.org/articles/10.3389/fcimb.2017.00064/full
Posted by: lindseysamot1957.blogspot.com
0 Response to "Label The Drawing Of An Enteric Bacterium With The Antigens And Virulence Factors."
Post a Comment